
When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report

 , , , , , and
, , , , , and 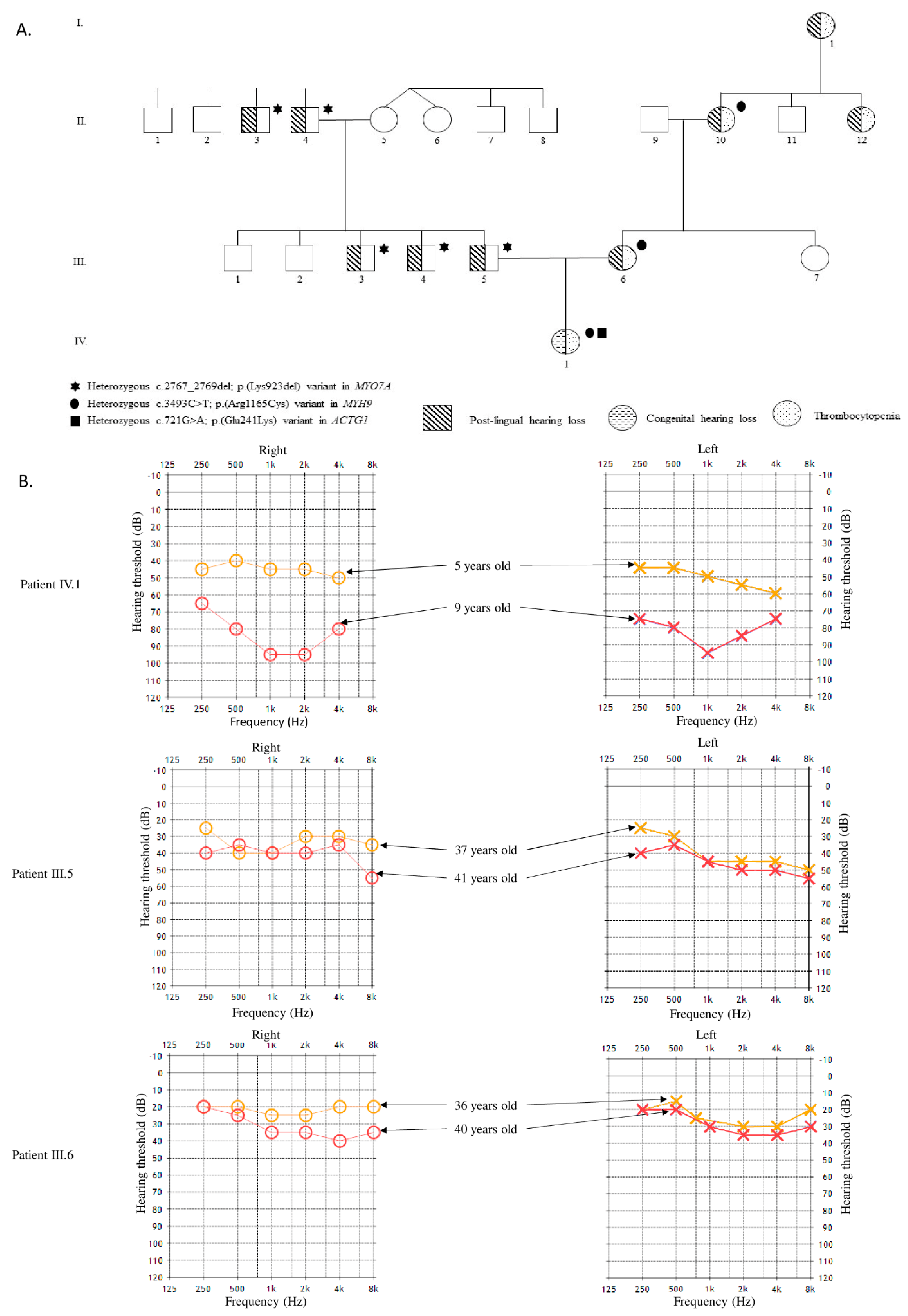{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Report
2.2. Molecular Analysis
2.2.1. NSHL Gene Panel Sequencing
2.2.2. Whole Exome Sequencing (WES)
3. Results
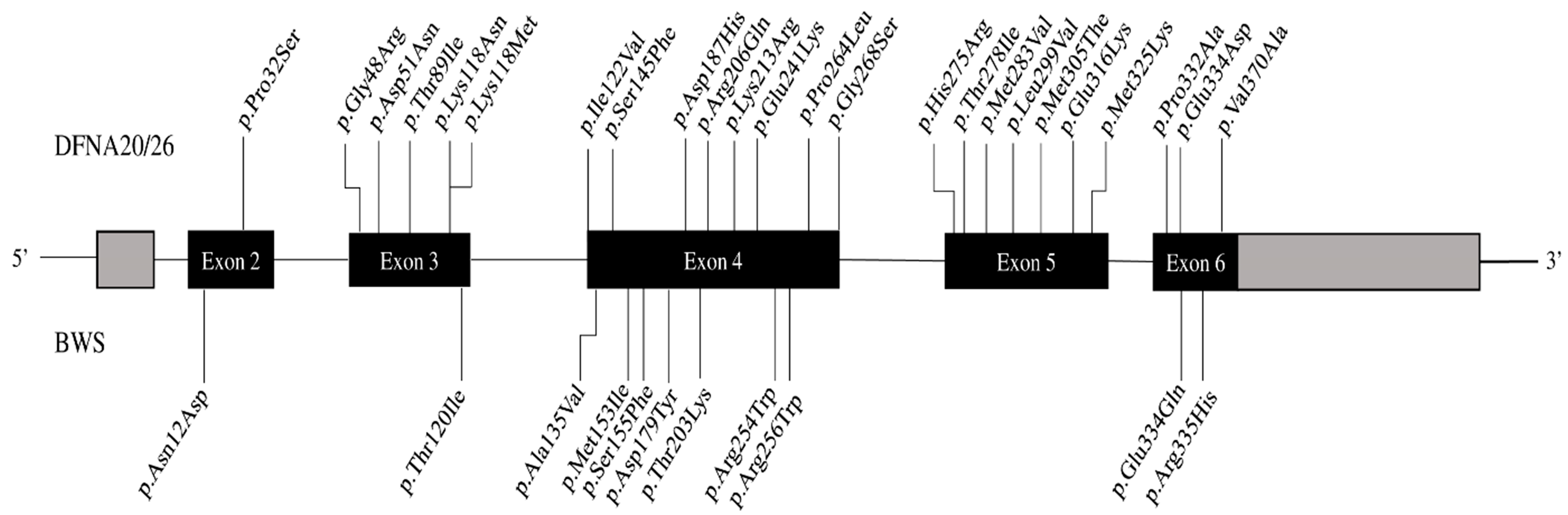
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, R.J.H.; Hildebrand, M.S.; Shearer, A.E.; Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Mirzaa, G.; Amemiya, A. Deafness and hereditary hearing loss overview. In GeneReviews; University of Washington: Seattle, WA, USA, 1999. [Google Scholar]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Blanco, K.; Sajan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A retrospective review of multiple findings in diagnostic exome sequencing: Half are distinct and half are overlapping diagnoses. Genet. Med. 2019, 21, 2199–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Yauy, K.; Baux, D.; Pegeot, H.; Van Goethem, C.; Mathieu, C.; Guignard, T.; Morales, R.J.; Lacourt, D.; Krahn, M.; Lehtokari, V.L.; et al. MoBiDiC prioritization algorithm, a free, accessible, and efficient pipeline for single-nucleotide variant annotation and prioritization for next-generation sequencing routine molecular diagnosis. J. Mol. Diagn. 2018, 20, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunishima, S.; Matsushita, T.; Kojima, T.; Amemiya, N.; Choi, Y.M.; Hosaka, N.; Inoue, M.; Jung, Y.; Mamiya, S.; Matsumoto, K.; et al. Identification of six novel MYH9 mutations and genotype-phenotype relationships in autosomal dominant macrothrombocytopenia with leukocyte inclusions. J. Hum. Genet. 2001, 46, 722–729. [Google Scholar] [CrossRef]
- Morín, M.; Bryan, K.E.; Mayo-Merino, F.; Goodyear, R.; Mencía, Á.; Modamio-Høybjør, S.; del Castillo, I.; Cabalka, J.M.; Richardson, G.; Moreno, F.; et al. In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum. Mol. Genet. 2009, 18, 3075–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Friedman, T.B.; Sellers, J.R.; Avraham, K.B. Unconventional myosins and the genetics of hearing loss. Am. J. Med. Genet. 1999, 89, 147–157. [Google Scholar] [CrossRef]
- Liu, X.Z.; Walsh, J.; Tamagawa, Y.; Kitamura, K.; Nishizawa, M.; Steel, K.P.; Brown, S.D. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 1997, 17, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Wolny, M.; Batchelor, M.; Knight, P.J.; Paci, E.; Dougan, L.; Peckham, M. Stable single alpha-helices are constant force springs in proteins. J. Biol. Chem. 2014, 289, 27825–27835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Jang, J.; Jin, H.S. A novel missense mutation in the ACTG1 gene in a family with congenital autosomal dominant deafness: A case report. Mol. Med. Rep. 2018, 17, 7611–7617. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, N.; Rump, A.; Koenig, R.; Der Kaloustian, V.M.; Halal, F.; Sonntag, K.; Krause, C.; Hackmann, K.; Hahn, G.; Schrock, E.; et al. Severe forms of Baraitser-Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur. J. Hum. Genet. 2014, 22, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venselaar, H.; Te Beek, T.; Kuipers, R.; Hekkelman, M.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivière, J.B.; Van Bon, B.W.; Hoischen, A.; Kholmanskikh, S.S.; O’Roak, B.J.; Gilissen, C.; Gijsen, S.; Sullivan, C.T.; Christian, S.L.; Abdul-Rahman, O.A.; et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012, 44, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainger, J.; Williamson, K.A.; Soares, D.C.; Truch, J.; Kurian, D.; Gillessen-Kaesbach, G.; Seawright, A.; Prendergast, J.; Halachev, M.; Wheeler, A.; et al. A recurrent de novo mutation in ACTG1 causes isolated ocular coloboma. Hum. Mutat. 2017, 38, 942–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cenni, C.; Mansard, L.; Blanchet, C.; Baux, D.; Vaché, C.; Baudoin, C.; Moclyn, M.; Faugère, V.; Mondain, M.; Jeziorski, E.; et al. When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report. Diagnostics 2021, 11, 1636. https://doi.org/10.3390/diagnostics11091636
Cenni C, Mansard L, Blanchet C, Baux D, Vaché C, Baudoin C, Moclyn M, Faugère V, Mondain M, Jeziorski E, et al. When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report. Diagnostics. 2021; 11(9):1636. https://doi.org/10.3390/diagnostics11091636
Chicago/Turabian StyleCenni, Camille, Luke Mansard, Catherine Blanchet, David Baux, Christel Vaché, Corinne Baudoin, Mélodie Moclyn, Valérie Faugère, Michel Mondain, Eric Jeziorski, and et al. 2021. "When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report" Diagnostics 11, no. 9: 1636. https://doi.org/10.3390/diagnostics11091636
APA StyleCenni, C., Mansard, L., Blanchet, C., Baux, D., Vaché, C., Baudoin, C., Moclyn, M., Faugère, V., Mondain, M., Jeziorski, E., Roux, A. -F., & Willems, M. (2021). When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report. Diagnostics, 11(9), 1636. https://doi.org/10.3390/diagnostics11091636