
A Novel Deletion Mutation of the F8 Gene for Hemophilia A

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical Characteristic and Laboratory Test
2.3. Sanger Sequencing
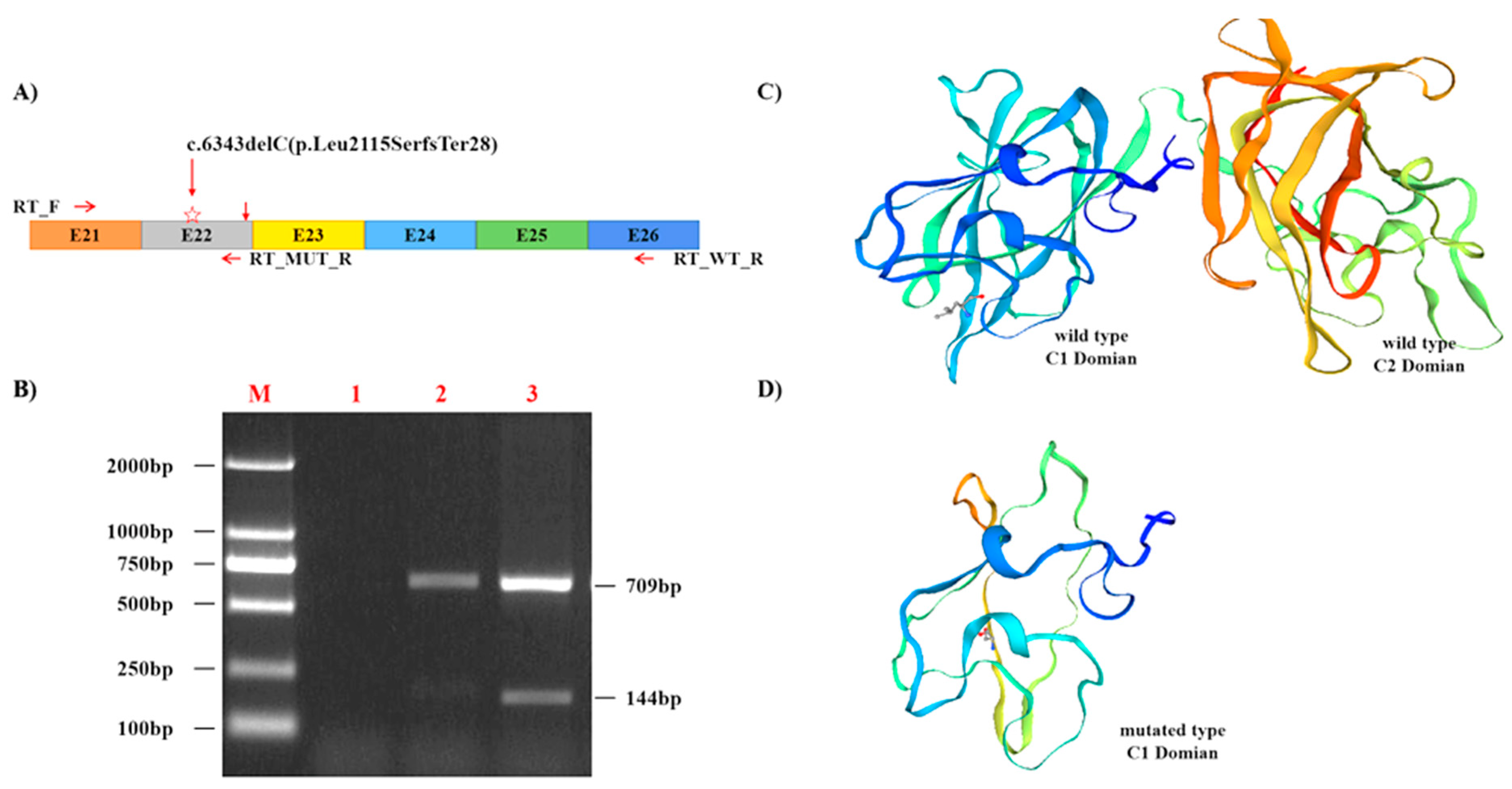
2.4. RNA and Reverse Transcription-PCR Analysis
2.5. Ethics
2.6. Data Analysis
3. Results
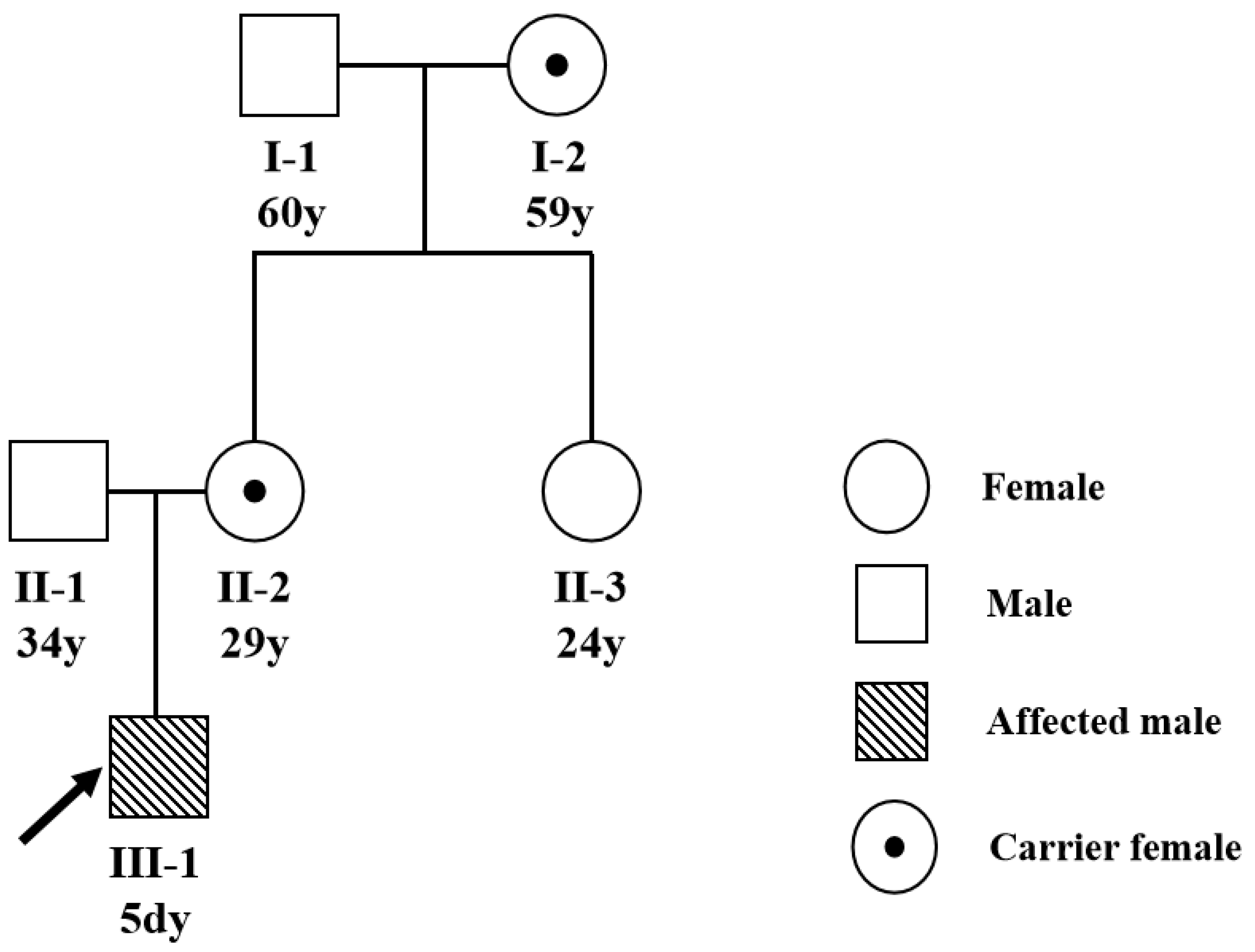
3.1. The Clinical Characteristics
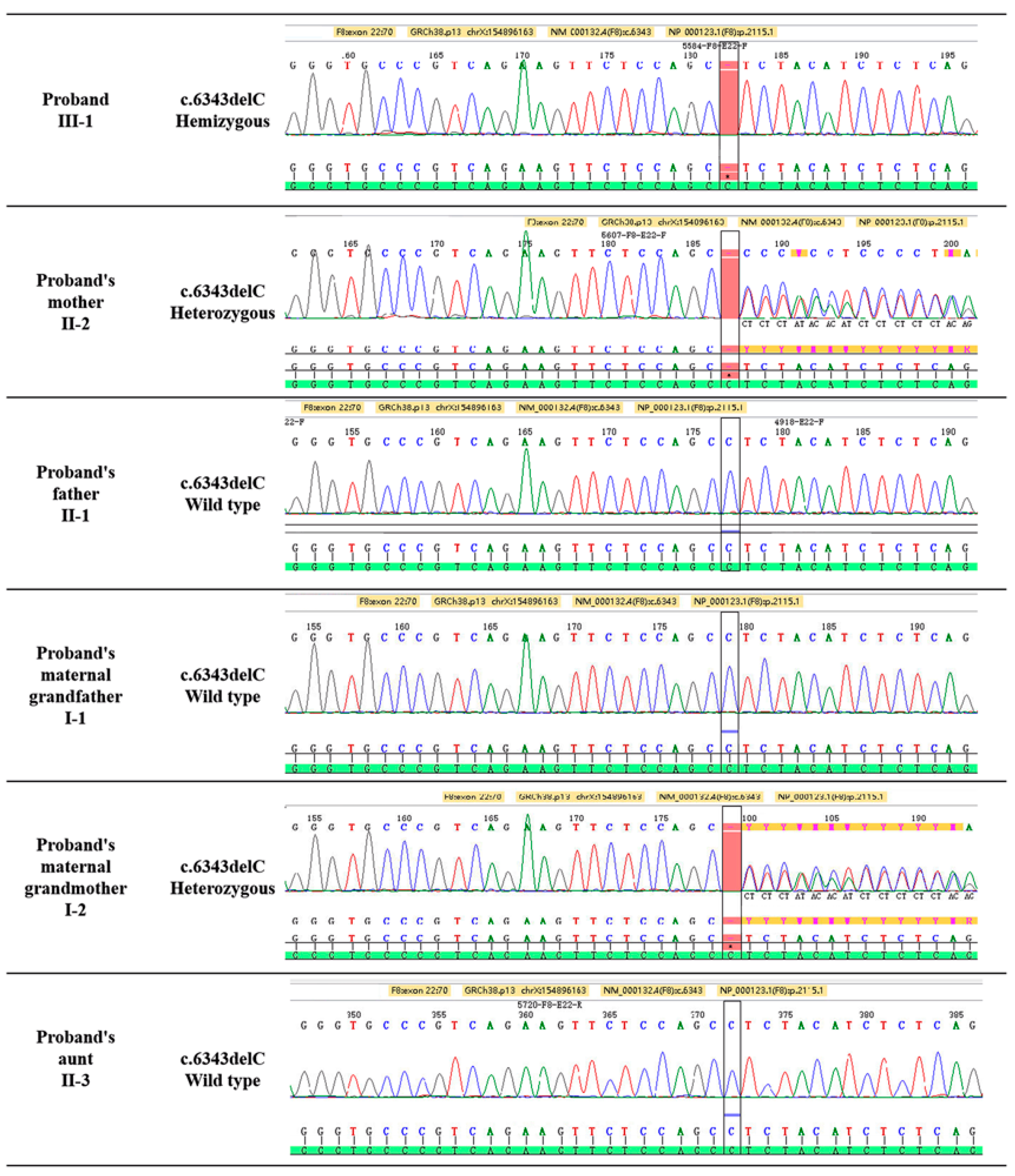
3.2. F8 Gene Mutation Detected by Sanger Sequencing
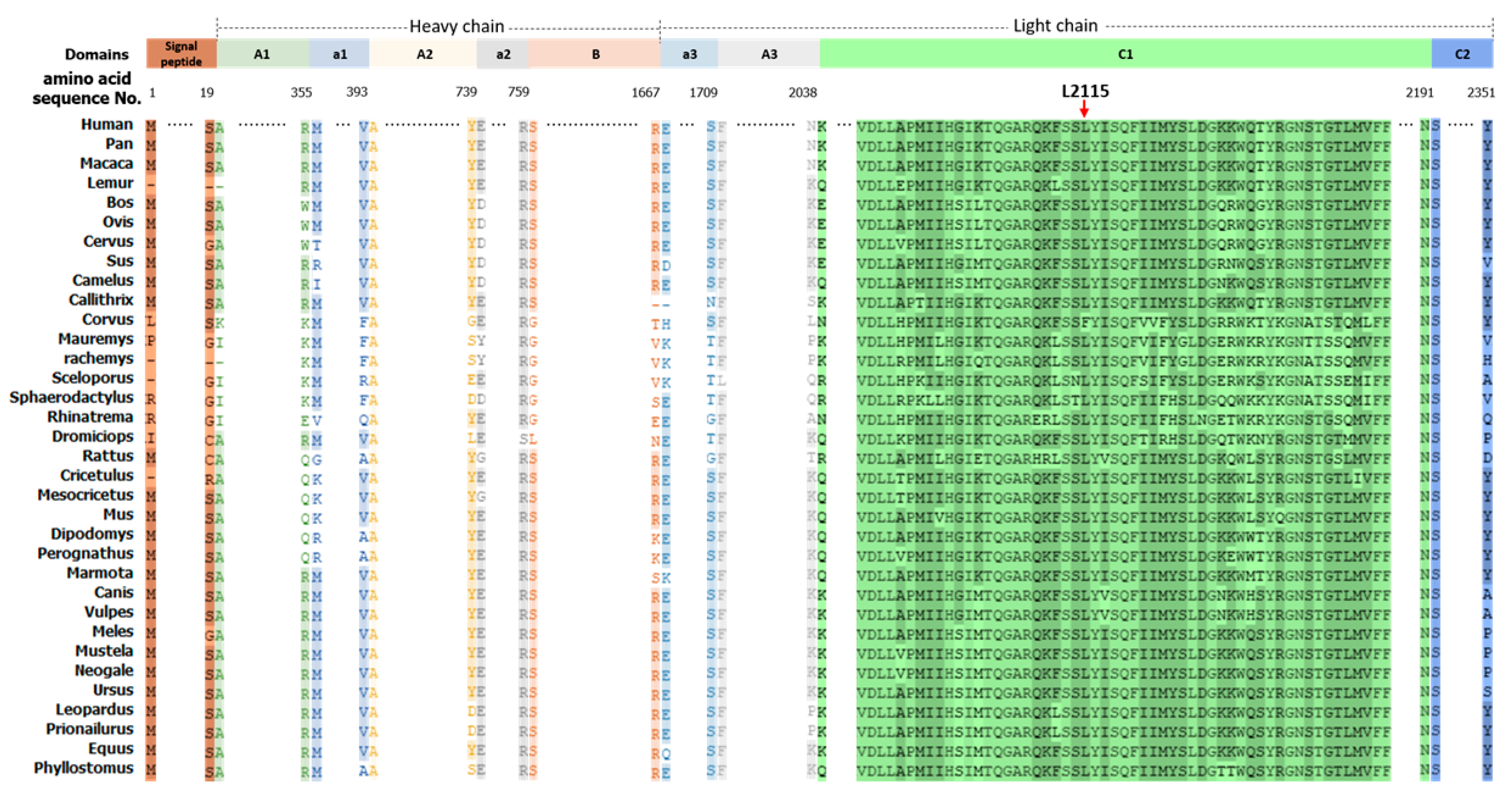
3.3. The F8 Mutation Is Pathogenic and Highly Conserved
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soucie, J.M.; Miller, C.H.; Dupervil, B.; Le, B.; Buckner, T.W. Occurrence rates of haemophilia among males in the United States based on surveillance conducted in specialized haemophilia treatment centres. Haemoph. Off. J. World Fed. Hemoph. 2020, 26, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lu, Y.; Ding, Q.; Wang, H.; Xi, X.; Wang, X. The status of carrier and prenatal diagnosis of haemophilia in China. Haemoph. Off. J. World Fed. Hemoph. 2012, 18, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Chistolini, A.; Papacchini, M.; Mazzucconi, M.G.; La Verde, G.; Arcieri, R.; Ferrari, A.; Paesano, R.; Pachi, A.; Mariani, G. Carrier detection and prenatal diagnosis in haemophilia A and B. Haematologica 1990, 75, 424–428. [Google Scholar] [PubMed]
- White, G.C., 2nd; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J.; Factor, V.; Factor, I.X.S. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar] [PubMed]
- Paroskie, A.; Gailani, D.; DeBaun, M.R.; Sidonio, R.F., Jr. A cross-sectional study of bleeding phenotype in haemophilia A carriers. Br. J. Haematol. 2015, 170, 223–228. [Google Scholar] [CrossRef]
- Stoilova-McPhie, S.; Lynch, G.C.; Ludtke, S.; Pettitt, B.M. Domain organization of membrane-bound factor VIII. Biopolymers 2013, 99, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Wang, X. The current situation of genetic diagnosis of hemophilia. J. Clin. Intern. Med. 2020, 37, 16–19. [Google Scholar] [CrossRef]
- Fuller, J.R.; Knockenhauer, K.E.; Leksa, N.C.; Peters, R.T.; Batchelor, J.D. Molecular determinants of the factor VIII/von Willebrand factor complex revealed by BIVV001 cryo-electron microscopy. Blood 2021, 137, 2970–2980. [Google Scholar] [CrossRef]
- McVey, J.H.; Rallapalli, P.M.; Kemball-Cook, G.; Hampshire, D.J.; Giansily-Blaizot, M.; Gomez, K.; Perkins, S.J.; Ludlam, C.A. The European Association for Haemophilia and Allied Disorders (EAHAD) Coagulation Factor Variant Databases: Important resources for haemostasis clinicians and researchers. Haemoph. Off. J. World Fed. Hemoph. 2020, 26, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Gouw, S.C.; van den Berg, H.M.; Oldenburg, J.; Astermark, J.; de Groot, P.G.; Margaglione, M.; Thompson, A.R.; van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef]
- Marder, V.J.; Aird, W.C.; Bennett, J.S.; Schulman, S.; White, G.C., II. Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 6th ed.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2012. [Google Scholar]
- Kaufman, R.J.; Powell, J.S. Molecular approaches for improved clotting factors for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Lin, L.; Yuan, C.; Nicolaes, G.A.; Chen, L.; Meehan, E.J.; Furie, B.; Furie, B.; Huang, M. Trp2313-His2315 of factor VIII C2 domain is involved in membrane binding: Structure of a complex between the C2 domain and an inhibitor of membrane binding. J. Biol. Chem. 2010, 285, 8824–8829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plug, I.; Mauser-Bunschoten, E.P.; Bröcker-Vriends, A.H.J.T.; van Amstel, H.K.P.; van der Bom, J.G.; van Diemen-Homan, J.E.M.; Willemse, J.; Rosendaal, F.R. Bleeding in carriers of hemophilia. Blood 2006, 108, 52–56. [Google Scholar] [CrossRef]
- Favier, R.; Lavergne, J.-M.; Costa, J.-M.; Caron, C.; Mazurier, C.; Viémont, M.; Delpech, M.; Valleix, S. Unbalanced X-chromosome inactivation with a novel FVIII gene mutation resulting in severe hemophilia A in a female. Blood 2000, 96, 4373–4375. [Google Scholar] [CrossRef]
- Aledort, L.; Mannucci, P.M.; Schramm, W.; Tarantino, M. Factor VIII replacement is still the standard of care in haemophilia A. Blood Transfus. 2019, 17, 479–486. [Google Scholar] [CrossRef]
- Fischer, K.; Astermark, J.; van der Bom, J.G.; Ljung, R.; Berntorp, E.; Grobbee, D.E.; van den Berg, H.M. Prophylactic treatment for severe haemophilia: Comparison of an intermediate-dose to a high-dose regimen. Haemoph. Off. J. World Fed. Hemoph. 2002, 8, 753–760. [Google Scholar] [CrossRef]
- Feldman, B.M.; Pai, M.; Rivard, G.E.; Israels, S.; Poon, M.C.; Demers, C.; Robinson, S.; Luke, K.H.; Wu, J.K.M.; Gill, K.; et al. Tailored prophylaxis in severe hemophilia A: Interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J. Thromb. Haemost. 2006, 4, 1228–1236. [Google Scholar] [CrossRef]
- Green, P.M.; Bagnall, R.D.; Waseem, N.H.; Giannelli, F. Haemophilia A mutations in the UK: Results of screening one-third of the population. Br. J. Haematol. 2008, 143, 115–128. [Google Scholar] [CrossRef]
- Chen, H.; Shi, M.; Gilam, A.; Zheng, Q.; Zhang, Y.; Afrikanova, I.; Li, J.; Gluzman, Z.; Jiang, R.; Kong, L.-J.; et al. Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing of human Factor VIII. Sci. Rep. 2019, 9, 16838. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Zhang, L.; Cheng, T.; Zhang, X. Research advances on gene therapy for hemophilia A. Zhonghua Xue Ye Xue Za Zhi 2015, 36, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.; Park, C.-Y.; Leem, J.W.; Cho, M.S.; Kim, D.-W. Restoration of FVIII expression by targeted gene insertion in the FVIII locus in hemophilia A patient-derived iPSCs. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zhou, M.; Wu, Y.; Li, Z.; Liu, X.; Wu, L.; Liang, D. ssODN-Mediated In-Frame Deletion with CRISPR/Cas9 Restores FVIII Function in Hemophilia A-Patient-Derived iPSCs and ECs. Mol. Ther. Nucleic Acids 2019, 17, 198–209. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Tm Value (°C) | GC Content (%) | Product Size (bp) |
|---|---|---|---|---|
| E1 | F: 5′-GGGTAAAGTTCCTTAAAATGCTCT-3′ R: 5′-TATGACGAAGAGAAGCAATGGACA-3′ | 57 60 | 37.5 41.67 | 536 |
| E2 | F: 5′-AAAGGATTTTTGGCTGATGCAGT-3′ R: 5′-TGGAGGTAAGCAGTTGGCATAG-3′ | 60 60 | 39.13 50 | 572 |
| E3 | F: 5′-ACTGCACCTCCAGTGCATAAG-3′ R: 5′-GTAACGCCACCATTACAAAGCA-3′ | 60 60 | 52.38 45.45 | 467 |
| E4 | F: 5′-ATGCAGAAAGTCCGTTTCTTATGT-3′ R: 5′-TCAGGTGAAGGAACACAAATGC-3′ | 59 59 | 37.5 45.45 | 422 |
| E5 | F: 5′-GAGACCTGACATCAAAGCCAAG-3′ R: 5′-TGAGGCCAGTCCGTTATCCA-3′ | 59 61 | 50 55 | 435 |
| E6 | F: 5′-F: 5′-TGAGAGGATAAGTGCTGTGTGC-3′ R: 5′-GATGCCGAGCTGTTTGTGAACT-3′ | 60 62 | 50 50 | 348 |
| E7 | F: 5′-ACGAATGAATGGTCAACAGGTGC-3′ R: 5′-GAAGCTGGAAACTAGGGGATCT-3′ | 62 59 | 47.83 50 | 628 |
| E8 | F: 5′-GGAAGGCCTAATAAGAGAGAAAGAA-3′ R: 5′-ATAGTCCCAGTCCTCCTCTTCA-3′ | 58 59 | 40 50 | 686 |
| E9 | F: 5′-CAAACCAACAAATCCTGAAGCCA-3′ R: 5′-GTGTCGAGTTTAGTGGGTGACATTA-3′ | 60 61 | 43.48 44 | 598 |
| E10 | F: 5′-GACCACAGTTTTCTTGTTGATCCT-3′ R: 5′-TGTCAGGCGACTCTTCACGA-3′ | 59 61 | 41.67 55 | 358 |
| E11 | F: 5′-AATAGGTGCGACTTTAGCTTCCA-3′ R: 5′-TCCATCCAGCAGGCACGTTT-3′ | 60 62 | 43.48 55 | 517 |
| E12 | F: 5′-GGATCAGTCACCCTCTTGTCC-3′ R: 5′-GGGTTATATGATCACGTGTGTTTGA-3′ | 60 59 | 57.14 40 | 545 |
| E13 | F: 5′-CCTGGGAATAAGATAATGGGCA-3′ R: 5′-ATCCCTGTACCTCAAGGAAGAAAA-3′ | 58 59 | 45.45 41.67 | 451 |
| E14_1 | F: 5′-AGGCATAGTACAACAGCAGCAA-3′ R: 5′-ATGAACTGGCATACTTGGGGG-3′ | 60 60 | 45.45 52.38 | 873 |
| E14_2 | F: 5′-TCCATCAGACAATTTGGCAGCAG-3′ R: 5′-GAGGCAAAACTACATTCTCTTGGA-3′ | 62 59 | 47.83 41.67 | 971 |
| E14_3 | F: 5′-CCAAGCAGCAGAAACCTATTTCTTA-3′ R: 5′-TTGGGCAAGTCTGGTTTCGG-3′ | 60 61 | 40.00 55.00 | 956 |
| E14_4 | F: 5′-AAGCAGTCATTTCTTACAAGGAGC-3′ R: 5′-TCATTGTTGGTGTCATCATCTGGTA-3′ | 60 60 | 41.67 40.00 | 948 |
| E15 | F: 5′-CAAAATGCTTCTCAGGCACCTA-3′ R: 5′-ATGTGCAAGGGACATTACCAA-3′ | 59 58 | 45.45 42.86 | 731 |
| E16 | F: 5′-TCTGTACCACTTCTTCCAGGGT-3′ R: 5′-CCATCCTCTTCAGTAGATTCCAGA-3′ | 60 59 | 50 45.83 | 524 |
| E17_E18 | F: 5′-TGGAATCTACTGAAGAGGATGGATT-3′ R: 5′-CACTGATTGTGTTCCCAGTGC-3′ | 59 60 | 40 52.38 | 847 |
| E19 | F: 5′-CCCCCAACTGTAAGGGTCAC-3′ R: 5′-CCTGACACAAGCAACCATTCC-3′ | 60 60 | 60 52.38 | 374 |
| E20 | F: 5′-GCATTTGTTGACGTTCTCCCAT-3′ R: 5′-GGAGAGGAGGAGATGTATTTGAGAGG-3′ | 60 62 | 45.45 50 | 314 |
| E21 | F: 5′-TGTTTTTCTCTATTTTCACCACAGC-3′ R: 5′-CCCCATATCTCTTTGTTCATGACTG-3′ | 59 59 | 36 44 | 364 |
| E22 | F: 5′-GGTGACTGCTTCACTTGCACA-3′ R: 5′-GAGCCTTGACACTACTACATTTTTG-3′ | 61 58 | 52.38 40 | 475 |
| E23 | F: 5′-CTTCACTTGCCCCAGACCTAAT-3′ R: 5′-CCCAGGACTATGCTGGTTTTAGC-3′ | 60 61 | 50 52.17 | 512 |
| E24 | F: 5’-TGCAAAAGTTAAAACCTGAGAAATG-3′ R: 5’-GTCTGCCCATAACCAAACTTCC-3′ | 57 60 | 32 50 | 422 |
| E25 | F: 5′-AGAGTGAGAAGTGCTGTGGTATGG-3′ R: 5′-AAAGTCACTGTGTTCTCTCAGAATG-3′ | 62 59 | 50 40 | 446 |
| E26 | F: 5′-TCCCAGATGCGTAGGACAGAGT-3′ R: 5′-AGCACAAAGGTAGAAGGCAAGC-3′ | 63 61 | 54.55 50 | 398 |
| RT_MUT | F: 5′-TGGATCAAGGTGGATCTGTTGG-3′ R: 5′-TCCTCGATAAGTCTGCCACT-3′ | 58 56 | 50.00 50.00 | 144 |
| RT_WT | F: 5′-TGGATCAAGGTGGATCTGTTGG-3′ R: 5′-GGTAGCGAGTCAGTAACGGTG-3′ | 58 58 | 50.00 57.14 | 709 |
| Test Items | Proband | Mother | Father | Aunt # | Grandmother # | Reference Ranges |
|---|---|---|---|---|---|---|
| RBC (1012 cells/L) | 2.43↓ | 4.28 | 4.52 | 4.50 | 3.70 | 3.6–6.6 (Children) 4.3–5.8 (Male) 3.8–5.1 (Female) |
| Hb (g/L) | 86↓ | 115 | 139 | 133 | 113 | 140–200 (Children) 130–175 (Male) 115–150 (Female) |
| PLT (109 cells/L) | 185↓ | 209 | 168 | 268 | 307 | 242–378 (Children) 125–350 (Adult) |
| PT (s) | 11.20 | 12.50 | 10.80 | 11.30 | 10.40 | 9–13 |
| APTT (s) | >170↑ | 36.80 | 28.7 | 29 | 37.80 | 20–40 |
| TT (s) | 15.40 | 16.30 | 17.30 | 14.30 | 15.90 | 14–21 |
| D-dimer (mg/L) | 6.26↑ | 0.51 | 0.18 | 0.32 | 0.48 | 0–0.55 |
| FDP (mg/L) | 15.26↑ | 1.80 | 1.50 | 2.07 | 2.26 | 0–5 |
| VWF:Ag | 61.90 | 65.30 | 103.20 | 71.80 | 123.50 | 50–200 |
| AT-III (%) | 44.80↓ | 85.30 | 94.80 | 82.70 | 94.40 | 80–120 |
| FVIII activity (%) | 2.0↓ | 75.40 | 154.30 | 116.50 | 69.70 | 60–189 |
| FIX activity (%) | 38.90↓ | 100.70 | 95.00 | 120.10 | 74.40 | 65–150 |
| FX activity (%) | 74.0↓ | 93.30 | 97.40 | 90.30 | 90.10 | 77–131 |
| FXI activity (%) | 67.60 | 100.70 | 71.70 | 101.70 | 81.20 | 65–150 |
| FXII activity (%) | 19.30↓ | 63.50 | 71.40 | 86.70 | 69.90 | 50–150 |
| G6PD enzyme activity (U/L) | 3491.4 | 3193.4 | 2954.3 | 2485.4 | 2103.5 | 2500–5800 (Newborn) 1700–4000 (Children) 1300–3600 (Adult) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Gu, J.; Chen, H.; Wu, Q.; Xiong, L.; Qiao, B.; Zhang, Y.; Xiao, H.; Tong, Y. A Novel Deletion Mutation of the F8 Gene for Hemophilia A. Diagnostics 2022, 12, 2876. https://doi.org/10.3390/diagnostics12112876
Wang J, Gu J, Chen H, Wu Q, Xiong L, Qiao B, Zhang Y, Xiao H, Tong Y. A Novel Deletion Mutation of the F8 Gene for Hemophilia A. Diagnostics. 2022; 12(11):2876. https://doi.org/10.3390/diagnostics12112876
Chicago/Turabian StyleWang, Jingwei, Jian Gu, Hongbing Chen, Qian Wu, Liang Xiong, Bin Qiao, Yan Zhang, Hongjun Xiao, and Yongqing Tong. 2022. "A Novel Deletion Mutation of the F8 Gene for Hemophilia A" Diagnostics 12, no. 11: 2876. https://doi.org/10.3390/diagnostics12112876
APA StyleWang, J., Gu, J., Chen, H., Wu, Q., Xiong, L., Qiao, B., Zhang, Y., Xiao, H., & Tong, Y. (2022). A Novel Deletion Mutation of the F8 Gene for Hemophilia A. Diagnostics, 12(11), 2876. https://doi.org/10.3390/diagnostics12112876