
The History and Future of Basic and Translational Cell-Free DNA Research at a Glance

{kind=link}
Abstract
:1. Introduction
2. Events in the Discovery of DNA Structure and Its Role in the Gene
3. Cytoplasmic DNA
4. DNA Mobility
5. Major Impact Studies
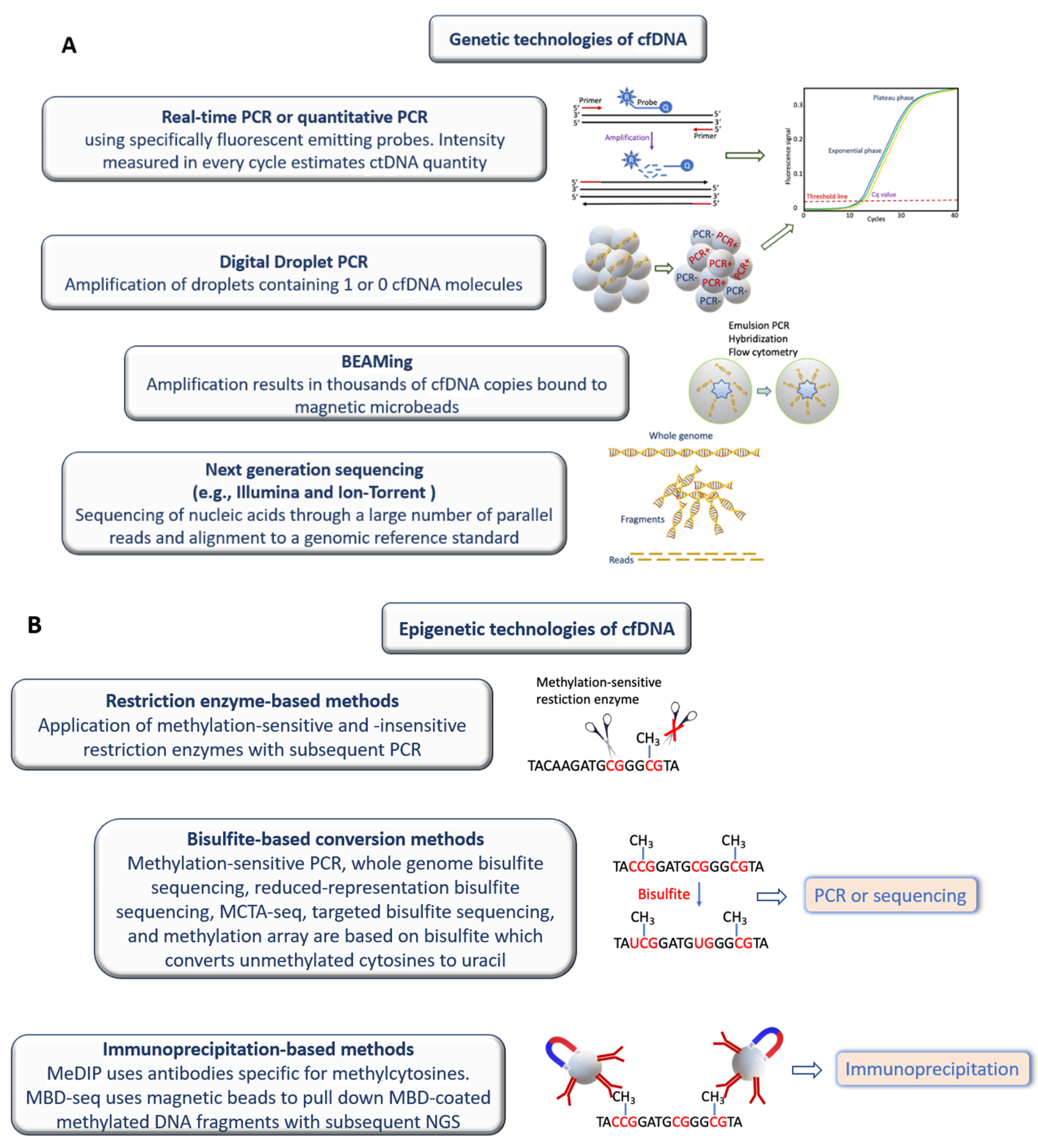
6. Methodological Development
7. Evolution of Sequencing
8. Polymerase Chain Reaction (PCR) Evolution
9. The Current Status of the Use of cfDNA
9.1. In Cancer
9.2. In General Medicine
10. Future of cfDNA
11. Ethical Considerations
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mandel, P. Les Acides Nucleiques Du Plasma Sanguin Chez l’ Homme [The Nucleic Acids in Blood Plasma in Humans]. CR Seances Soc. Biol.Fil. 1948, 142, 241–243. [Google Scholar]
- Koffler, D.; Agnello, V.; Winchester, R.; Kunkel, H.G. The Occurrence of Single-Stranded DNA in the Serum of Patients with Systemic Lupus Erythematosus and Other Diseases. J. Clin. Investig. 1973, 52, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahan, P.B. A Brief History and the Present and Future Status of CNAPS. In Circulating Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring: An Introduction; Gahan, P.B., Ed.; Springer Science + Business Media: Dordrecht, The Netherlands, 2015; p. 471. [Google Scholar]
- Stroun, M.; Anker, P.; Maurice, P.; Gahan, P.B. Circulating Nucleic Acids in Higher Organisms. Int. Rev. Cytol. 1977, 51, 1–48. [Google Scholar] [CrossRef]
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type Iii. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef]
- Brachet, J. The Use of Basic Dyes and Ribonuclease for the Cytochemical Detection of Ribonucleic Acid. J. Cell Sci. 1953, 3, 1–10. [Google Scholar] [CrossRef]
- Caspersson, T.; Schultz, J. Ribonucleic Acids in Both Nucleus and Cytoplasm, and the Function of the Nucleolus. Proc. Natl. Acad. Sci. USA 1940, 26, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Alfert, M.; Swift, H. Nuclear DNA constancy: A critical evaluation of some exceptions reported by lison and pasteels. Exp. Cell Res. 1953, 5, 455–460. [Google Scholar] [CrossRef]
- Chargaff, E. Chemical specificity of nucleic acids and mechanism of their enzymatic degradation. Experientia 1950, 6, 201–209. [Google Scholar] [CrossRef]
- Broomhead, J.M. The structures of pyrimidines and purines. IV. The crystal structure of guanine hydrochloride and its relation to that of adenine hydrochloride. Acta Crystallogr. 1951, 4, 92–100. [Google Scholar] [CrossRef]
- Broomhead, J.M. The structure of pyrimidines and purines. II. A determination of the structure of adenine hydrochloride by X-ray methods. Acta Crystallogr. 1948, 1, 324–329. [Google Scholar] [CrossRef]
- Wilkins, M.H.F.; Stokes, A.R.; Wilson, H.R. Molecular Structure of Nucleic Acids: Molecular Structure of Deoxypentose Nucleic Acids. Nature 1953, 171, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Pelc, S. Nuclear incorporation of P32 as demonstrated by autoradiographs. Exp. Cell Res. 1951, 2, 178–187. [Google Scholar] [CrossRef]
- Chayen, J.; Denby, E. The distribution of deoxyribonucleic acid in homogenates of plant roots. Exp. Cell Res. 1960, 20, 182–197. [Google Scholar] [CrossRef]
- Srb, A.M.; Owen, R.D. General Genetics; W.H. Freeman & Co.: San Francisco, CA, USA, 1958. [Google Scholar]
- Gahan, P.B.; Chayen, J.; Silcox, A.A. Cytoplasmic Localization of Deoxyribonucleic Acid in Allium cepa. Nature 1962, 195, 1115–1116. [Google Scholar] [CrossRef] [PubMed]
- Chevremont, M.; Bassleer, R.; Baeckeland, E. New research on desoxyribonucleic acids in cooled, then reheated, cultures of fibroblasts. Cytophotometric and histoautoradiographic studies. Cytoplasmic localization of DNA. Arch. Biol. 1961, 72, 501–524. [Google Scholar]
- Sonneborn, T.M. Beyond the Gene. Am. Sci. 1949, 37, e1231. [Google Scholar]
- Stroun, M.; Mathon, C.; Stroun, J. Modifications Transmitted to the Offspring Provoked by Heterograft in Solanum melongena. Arch. Sci. 1963, 16, 39. [Google Scholar]
- Strou, N.; Stroun-Guttière, S.; Ross, I. Transfer to the progeny of alterations induced in the White Leghorn by the repeated injections of heterologous blood. Transplantation 1964, 2, 446. [Google Scholar] [CrossRef]
- Glouchtchenko, I.E. Vegetative Hybridization in Massive Horizontal Gene Transfer in Plants; Academy Nauk SSR: Moscow, Russia, 1948; 240p. (In Russian) [Google Scholar]
- Stroun, M.; Mathon, C.C.; Stroun, J. Alteration of Hereditary Traits in Solanum melongena Induced by Grafts with Solanum Nigrum. In Proceedings of the 11th International Congress of Genetics (la Haye), The Hague, The Netherlands, 1 September 1963; pp. 1–218. [Google Scholar]
- Hirata, Y. Graft-Induced Changes in Eggplant (S. melongena L.) I. Appearance of the Changes. Euphytica 1986, 35, 395–401. [Google Scholar] [CrossRef]
- Yagishita, N. Studies on Graft Hybrids of Capsicum annuum L. II. J. Plant Res. 1961, 74, 480–489. [Google Scholar] [CrossRef]
- Yagishita, N. Studies on Graft Hybrids of Capsicum annuum L. J. Plant Res. 1961, 74, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Ohta, Y.; Van Chuong, P. Hereditary changes in Capsicum annuum L. I. Induced by ordinary grafting. Euphytica 1975, 24, 355–368. [Google Scholar] [CrossRef]
- Frankel, R. Further evidence on graft induced transmission to progeny of cytoplasmic male sterility in petunia. Genetics 1962, 47, 641–646. [Google Scholar] [CrossRef]
- Gahan, P. Circulating nucleic acids: Possible inherited effects. Biol. J. Linn. Soc. 2013, 110, 931–948. [Google Scholar] [CrossRef] [Green Version]
- Michurin, I.V. Selected Works; Foreign Languages Publishing House: Moscow, Russia, 1949. [Google Scholar]
- Liu, Y. Historical and Modern Genetics of Plant Graft Hybridization. Adv. Genet. 2006, 56, 101–129. [Google Scholar] [CrossRef]
- Leroy, P. Divergence between the Phenotypes of Adult Hybrids Obtained by Crossing Rhode Island Red M44 or Modified Rhode Island Red Males with White Wyndotte Females. CR Acad. Sci. 1968, 266, 516–518. [Google Scholar]
- Leroy, P.; Benoit, J. Comparison between Descendants of Rhode Island Red M44 Pullets Injected with Whole Blood or the Erythrocyte Nuclear Complex of the Guinea Fowl. CR Acad. Sci. 1966, 262, 805–808. [Google Scholar]
- Leroy, P.; Benoit, J. Results Obtained with Third and Fourth Generation Progeny of Rhode Island Red Fowls Treated with Guinea Fowl Blood. CR Acad. Sci. 1963, 256, 4501–4504. [Google Scholar]
- Leroy, P.; Venderly, R.; Benoit, J.; Venderly, C. Divergences Observed in the Descendants of Rhode Island Red Chickens M-44 after Injections of Specifically Different Bloods or Fractions of Blood. In Proceedings of the The Thirteenth World’s Poultry Congress Proceedings, Kiev, Ukraine, 22 January 1966. [Google Scholar]
- Leroy, P.; Benoit, J.; Venderly, R.; Venderly, C. The Effects of Injection of Nuclear Substances from Guinea-Fowl Erythrocytes on Rhode Island Red of Known Genotype. CR Acad. Sci. 1964, 258, 1905–1907. [Google Scholar]
- Liu, Y. A new perspective on Darwin’s Pangenesis. Biol. Rev. 2008, 83, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, L.; Brown, J.; Charles, P.; Huart, R.; Remy, J.; Jacobs, M.; Watters, C. Fate of Exogenous DNA in Mammals and Plants. In Proceedings of the Workshop on Mechanisms and Prospects of Genetic Exchange, Berlin, Germany, 11–13 December 1971. [Google Scholar]
- Gahan, P.B.; Anker, P.; Stroun, M. An Autoradiographic Study of Bacterial DNA in Lycopersicon esculentum. Ann. Bot. 1973, 37, 681–685. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Charles, P.; Ledoux, L. Translocation of DNA of Bacterial Origin in Lycopersicum esculentum by Ul-tracentrifugation in Caesium Chloride Gradient. Nature 1967, 215, 975–976. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Anker, P.; Charles, P.; Ledoux, L. Fate of Bacterial Deoxyribonucleic Acid in Lycopersicon esculentum. Nature 1966, 212, 397–398. [Google Scholar] [CrossRef]
- Gahan, P.B.; Anker, P.; Stroun, M.; Jacob, K. DNA-Induced Chromosome Damage inVicia Faba. Caryologia 1969, 22, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Stroun, M. Mon cher collègue je ne serai pas recteur. In Une Aventure Dans Le Monde de Université et de La Recherche Scientifique Suisse; Editions L’Harmattan: Paris, France, 2011. [Google Scholar]
- Harding, C.V.; Heuser, J.E.; Stahl, P.D. Exosomes: Looking Back Three Decades and into the Future. J. Cell Biol. 2013, 200, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2014, 34, 474–490. [Google Scholar] [CrossRef]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [Green Version]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Weidling, I.; Koppel, S.; Menta, B.; Ortiz, J.P.; Kalani, A.; Wilkins, H.M.; Swerdlow, R.H. Detection of mitochondria-pertinent components in exosomes. Mitochondrion 2020, 55, 100–110. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, J.; Miao, Y.; Zhang, Q. The effect of extracellular vesicles on the regulation of mitochondria under hypoxia. Cell Death Dis. 2021, 12, 358. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic Characteristics of the DNA Found in the Plasma of Cancer Patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Lyautey, J.; Lederrey, C.; Maurice, P.A. Isolation and characterization of DNA from the plasma of cancer patients. Eur. J. Cancer Clin. Oncol. 1987, 23, 707–712. [Google Scholar] [CrossRef]
- Lo, D.; Sargent, I.L.; Redman, C.W.; Lo, Y.M.D.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.G. Presence of Fetal DNA in Maternal Plasma and Serum Early Report Presence of Fetal DNA in Maternal Plasma and Serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Lo, Y.-M.; Bowell, P.; Selinger, M.; Mackenzie, I.; Chamberlain, P.; Gillmer, M.; Littlewood, T.; Fleming, K.; Wainscoat, J. Prenatal determination of fetal RhD status by analysis of peripheral blood of rhesus negative mothers. Lancet 1993, 341, 1147–1148. [Google Scholar] [CrossRef]
- Perrot, A.; Horn, R. The ethical landscape(s) of non-invasive prenatal testing in England, France and Germany: Findings from a comparative literature review. Eur. J. Hum. Genet. 2021, 1–6. [Google Scholar] [CrossRef]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef]
- Schwarzenbach, H. CNAPS and General Medicine: Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring. An Introduction; Gahan, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 5. [Google Scholar]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C.C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Kaiser, A. Structure and base sequence in the cohesive ends of bacteriophage lambda DNA. J. Mol. Biol. 1968, 35, 523–537. [Google Scholar] [CrossRef]
- Kelly, T.J.; Smith, H.O. A restriction enzyme from Hemophilus influenzae: II. Base sequence of the recognition site. J. Mol. Biol. 1970, 51, 393–409. [Google Scholar] [CrossRef]
- Smith, H.O.; Welcox, K. A Restriction enzyme from Hemophilus influenzae: I. Purification and general properties. J. Mol. Biol. 1970, 51, 379–391. [Google Scholar] [CrossRef]
- Hutchison, C.A. DNA sequencing: Bench to bedside and beyond. Nucleic Acids Res. 2007, 35, 6227–6237. [Google Scholar] [CrossRef]
- Gilbert, W.; Maxam, A. The Nucleotide Sequence of the lac Operator. Proc. Natl. Acad. Sci. USA 1973, 70, 3581–3584. [Google Scholar] [CrossRef] [Green Version]
- Maniatis, T.; Ptashne, M.; Barrell, B.G.; Donelson, J. Sequence of a represser-binding site in the DNA of bacteriophage λ. Nature 1974, 250, 394–397. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, C.A.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucliotide Sequence of Bacteriophage Phi X174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.H.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921, Correction in Nature 2001, 412, 565–566; Erratum in Nature 2001, 411, 720. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S. Frederick Sanger (1918–2013). Science 2014, 343, 262. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Rogers, Y.H.; Venter, J.C. Genomics: Massively Parallel Sequencing. Nature 2005, 437, 326–327. [Google Scholar] [CrossRef]
- Lavebratt, C.; Şengül, S. Single nucleotide polymorphism (SNP) allele frequency estimation in DNA pools using Pyrosequencing™. Nat. Protoc. 2006, 1, 2573–2582. [Google Scholar] [CrossRef]
- Ronaghi, M.; Karamohamed, S.; Pettersson, B.; Uhlen, M.; Nyrén, P. Real-Time DNA Sequencing Using Detection of Pyrophosphate Release. Anal. Biochem. 1996, 242, 84–89. [Google Scholar] [CrossRef]
- Bennett, S.T.; Barnes, C.L.; Cox, A.; Davies, L.; Brown, C. Toward the 1000 Dollars Human Genome. Pharmacogenomics 2005, 6. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2015, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Galibert, F.; Netter, P. Tribute to Kary Mullis. Bull. L’academie Natl. Med. 2021, 16, 205. [Google Scholar]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R.H. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gunter, L.E.; Kochert, G.; Giannasi, D.E. Phylogenetic relationships of theJuglandaceae. Oesterreichische Bot. Z. 1994, 192, 11–29. [Google Scholar] [CrossRef]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [Green Version]
- Morley, A.A. Digital PCR: A Brief History. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Madic, J.; Zocevic, A.; Senlis, V.; Fradet, E.; Andre, B.; Muller, S.; Dangla, R.; Droniou, M. Three-color crystal digital PCR. Biomol. Detect. Quantif. 2016, 10, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, H.; Xu, Y.; Tureckova, A.; Zahradník, P.; Chang, H.; Neuzil, P. Versatile digital polymerase chain reaction chip design, fabrication, and image processing. Sens. Actuators B Chem. 2018, 283, 677–684. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [Green Version]
- Toldrà, A.; Rubio, M.J.; Andree, K.B.; Fernández-Tejedor, M.; Diogène, J.; Katakis, I.; O’Sullivan, C.K.; Campàs, M. Detection and quantification of the toxic marine microalgae Karlodinium veneficum and Karlodinium armiger using recombinase polymerase amplification and enzyme-linked oligonucleotide assay. Anal. Chim. Acta 2018, 1039, 140–148. [Google Scholar] [CrossRef]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain reaction product by utilizing the 5’----3’ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, G.V.F.; Lima, J.S.; De Oliveira, A.C.D.S.; Da Silva, J.B.; Roos, T.B.; De Moraes, C.M. SYBR Green qPCR Technique for the Detection of Trypanosoma cruzi in Açaí Pulp. Foodborne Pathog. Dis. 2020, 17, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunt, M.; Hillebrand, T.; Schwarzenbach, H. Clinical relevance of size selection of circulating DNA. Transl. Cancer Res. 2018, 7, S171–S184. [Google Scholar] [CrossRef]
- Schwarzenbach, H. Circulating nucleic acids as biomarkers in breast cancer. Breast Cancer Res. 2013, 15, 211. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damodaran, S.; Berger, M.F.; Roychowdhury, S. Clinical tumor sequencing: Opportunities and challenges for precision cancer medicine. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e175–e182. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.-J.; Jardin, F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals 2021, 14, 596. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.; Kopetz, S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Szilágyi, M.; Pös, O.; Márton, É.; Buglyó, G.; Soltész, B.; Keserű, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Kodahl, A.R.; Ehmsen, S.; Pallisgaard, N.; Jylling, A.M.B.; Jensen, J.D.; Laenkholm, A.-V.; Knoop, A.; Ditzel, H.J. Correlation between circulating cell-free PIK 3 CA tumor DNA levels and treatment response in patients with PIK 3 CA-mutated metastatic breast cancer. Mol. Oncol. 2018, 12, 925–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [Green Version]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.C.A.; Woo, J.K.S.; King, A.; Zee, B.C.-Y.; Lam, W.K.J.; Chan, S.L.; Chu, S.W.I.; Mak, C.; Tse, I.O.L.; Leung, S.Y.M.; et al. Analysis of Plasma Epstein–Barr Virus DNA to Screen for Nasopharyngeal Cancer. N. Engl. J. Med. 2017, 377, 513–522. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Park, B.H. Circulating Tumor DNA as a Marker for Disease Relapse in Early-Stage Breast Cancer—Bad Blood. JAMA Oncol. 2019, 5, 1479. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- De Vlaminck, I.; Martin, L.; Kertesz, M.; Patel, K.; Kowarsky, M.; Strehl, C.; Cohen, G.; Luikart, H.; Neff, N.F.; Okamoto, J.; et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc. Natl. Acad. Sci. USA 2015, 112, 13336–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vlaminck, I.; Valantine, H.A.; Snyder, T.M.; Strehl, C.; Cohen, G.; Luikart, H.; Neff, N.F.; Okamoto, J.; Bernstein, D.; Weisshaar, D.; et al. Circulating Cell-Free DNA Enables Noninvasive Diagnosis of Heart Transplant Rejection. Sci. Transl. Med. 2014, 6, 241ra77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solakoglu, Ö.; Steinbach, B.; Götz, W.; Heydecke, G.; Pantel, K.; Schwarzenbach, H. Characterization of circulating DNA in plasma of patients after allogeneic bone grafting. Clin. Oral Investig. 2019, 23, 4243–4253. [Google Scholar] [CrossRef] [Green Version]
- Chiu, R.W.K.; Chan, K.C.A.; Gao, Y.; Lau, V.Y.M.; Zheng, W.; Leung, T.Y.; Foo, C.H.F.; Xie, B.; Tsui, N.B.Y.; Lun, F.M.F.; et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Batey, A.; Struble, C.; Musci, T.; Song, K.; Oliphant, A. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat. Diagn. 2013, 33, 662–666. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [Green Version]
- Polina, I.A.; Ilatovskaya, D.V.; DeLeon-Pennell, K.Y. Cell free DNA as a diagnostic and prognostic marker for cardiovascular diseases. Clin. Chim. Acta 2020, 503, 145–150. [Google Scholar] [CrossRef]
- Khachatoorian, Y.; Khachadourian, V.; Chang, E.; Sernas, E.R.; Reed, E.F.; Deng, M.; Piening, B.D.; Pereira, A.C.; Keating, B.; Cadeiras, M. Noninvasive biomarkers for prediction and diagnosis of heart transplantation rejection. Transplant. Rev. 2020, 35, 100590. [Google Scholar] [CrossRef]
- Kahroba, H.; Ramezani, B.; Maadi, H.; Sadeghi, M.R.; Jaberie, H.; Ramezani, F. The role of Nrf2 in neural stem/progenitors cells: From maintaining stemness and self-renewal to promoting differentiation capability and facilitating therapeutic application in neurodegenerative disease. Ageing Res. Rev. 2020, 65, 101211. [Google Scholar] [CrossRef]
- Ozturk, E.A.; Caner, A. Liquid Biopsy for Promising Non-invasive Diagnostic Biomarkers in Parasitic Infections. Acta Parasitol. 2021, 67, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, T.I.; Hill, M.; Drury, S.; Mason, S.; Jenkins, L.; Morris, S.; Chitty, L.S. Non-invasive prenatal diagnosis (NIPD) for single gene disorders: Cost analysis of NIPD and invasive testing pathways. Prenat. Diagn. 2016, 36, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessies, D.C.; Greuter, M.J.; van Rooijen, K.L.; Linders, T.C.; Lanfermeijer, M.; Ramkisoensing, K.L.; Grijseels, F.E.; van Doorn, B.; Meijer, G.A.; Koopman, M.; et al. Abstract 2276: Performance and Cost Comparison of Circulating Tumor DNA Detection Platforms. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Hellwig, S.; Nix, D.A.; Gligorich, K.M.; O’Shea, J.M.; Thomas, A.; Fuertes, C.L.; Bhetariya, P.J.; Marth, G.T.; Bronner, M.P.; Underhill, H.R. Automated size selection for short cell-free DNA fragments enriches for circulating tumor DNA and improves error correction during next generation sequencing. PLoS ONE 2018, 13, e0197333. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Forman, M.; Hulme, R.; Old, R.W.; Ryan, D.; Mazey, R.; Risley, M.D. The IONA® Test: Development of an Automated Cell-Free DNA-Based Screening Test for Fetal Trisomies 13, 18, and 21 That Employs the Ion Proton Semiconductor Sequencing Platform. Fetal Diagn. Ther. 2017, 42, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, O.; Ahola, T.; Dahl, F.; Karlsson, F.; Persson, F.; Karlberg, O.; Roos, F.; Alftrén, I.; Andersson, B.; Barkenäs, E.; et al. Clinical validation of a novel automated cell-free DNA screening assay for trisomies 21, 13, and 18 in maternal plasma. Prenat. Diagn. 2019, 39, 1011–1015. [Google Scholar] [CrossRef]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.-J.; Crispin-Ortuzar, M.; Chin, S.-F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2021, 601, 623–629. [Google Scholar] [CrossRef]
- Ko, J.; Baldassano, S.N.; Loh, P.-L.; Kording, K.; Litt, B.; Issadore, D. Machine learning to detect signatures of disease in liquid biopsies—A user’s guide. Lab Chip 2017, 18, 395–405. [Google Scholar] [CrossRef]
- Lewis, C.; Chitty, L.S. Societal Aspects: Ethics. In Circulating Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring; Gahan, P.B., Ed.; Springer: Dordrecht, The Netherlands; Berlin/Heidelberg, Germany; New York, NY, USA; London, UK, 2014; pp. 381–398. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gahan, P.B.; Schwarzenbach, H.; Anker, P. The History and Future of Basic and Translational Cell-Free DNA Research at a Glance. Diagnostics 2022, 12, 1192. https://doi.org/10.3390/diagnostics12051192
Gahan PB, Schwarzenbach H, Anker P. The History and Future of Basic and Translational Cell-Free DNA Research at a Glance. Diagnostics. 2022; 12(5):1192. https://doi.org/10.3390/diagnostics12051192
Chicago/Turabian StyleGahan, Peter B., Heidi Schwarzenbach, and Philippe Anker. 2022. "The History and Future of Basic and Translational Cell-Free DNA Research at a Glance" Diagnostics 12, no. 5: 1192. https://doi.org/10.3390/diagnostics12051192
APA StyleGahan, P. B., Schwarzenbach, H., & Anker, P. (2022). The History and Future of Basic and Translational Cell-Free DNA Research at a Glance. Diagnostics, 12(5), 1192. https://doi.org/10.3390/diagnostics12051192