
Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cohort
2.2. Laboratory Methods
2.2.1. RNA Extraction
2.2.2. Discovery Study
Microarrays
qPCR
2.2.3. Validation Study
qRT-PCR Assays
2.3. Statistical Analysis
2.3.1. Validation Analysis
2.3.2. Data Preprocessing
2.3.3. Differential Expression
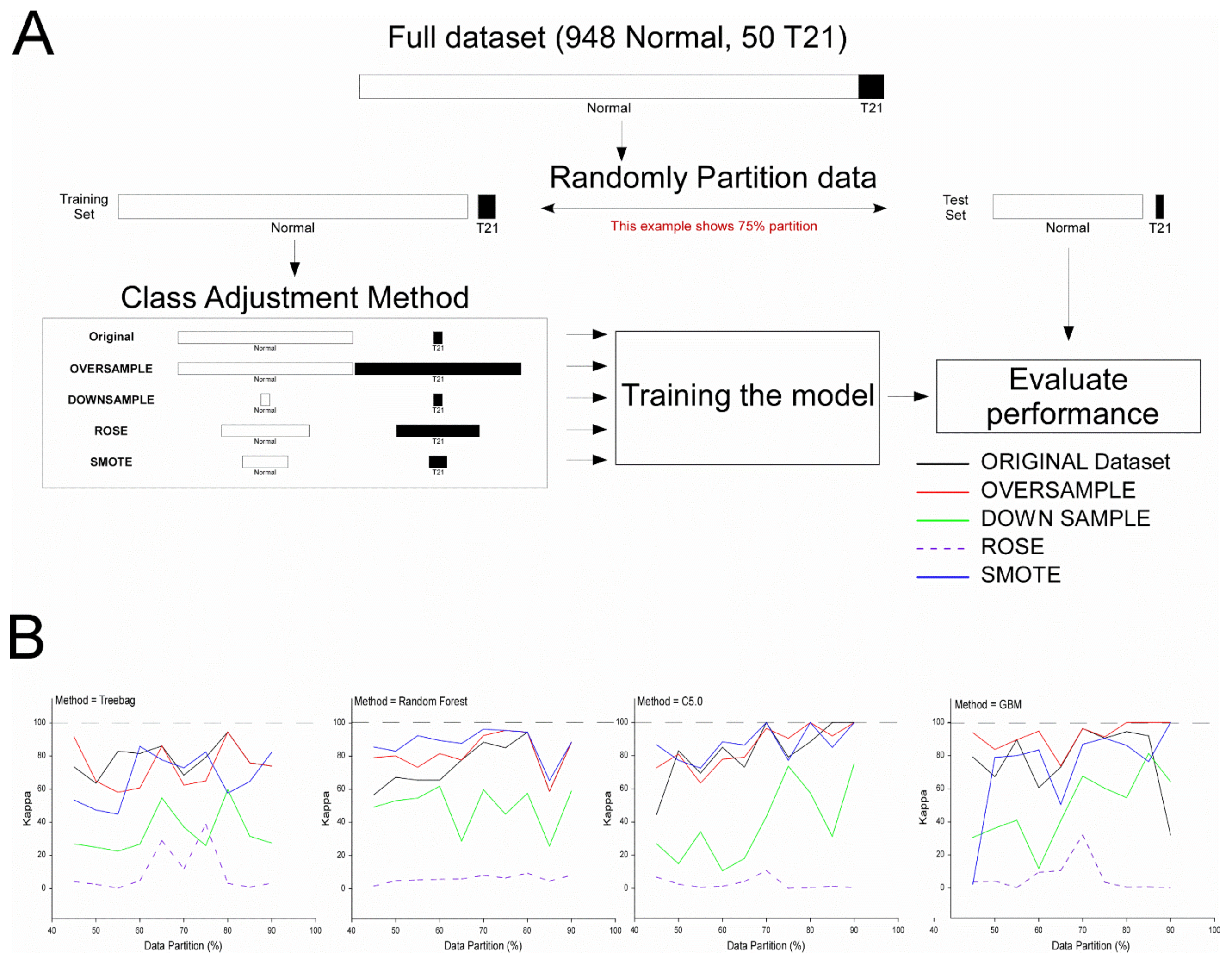
2.3.4. Machine Learning
3. Results
3.1. Discovery
3.1.1. Study Subjects
3.1.2. Focused Search Following Effect Size Stratification and Removal of RNAs Affected by Race
3.2. Validation Study
3.2.1. Study Subjects
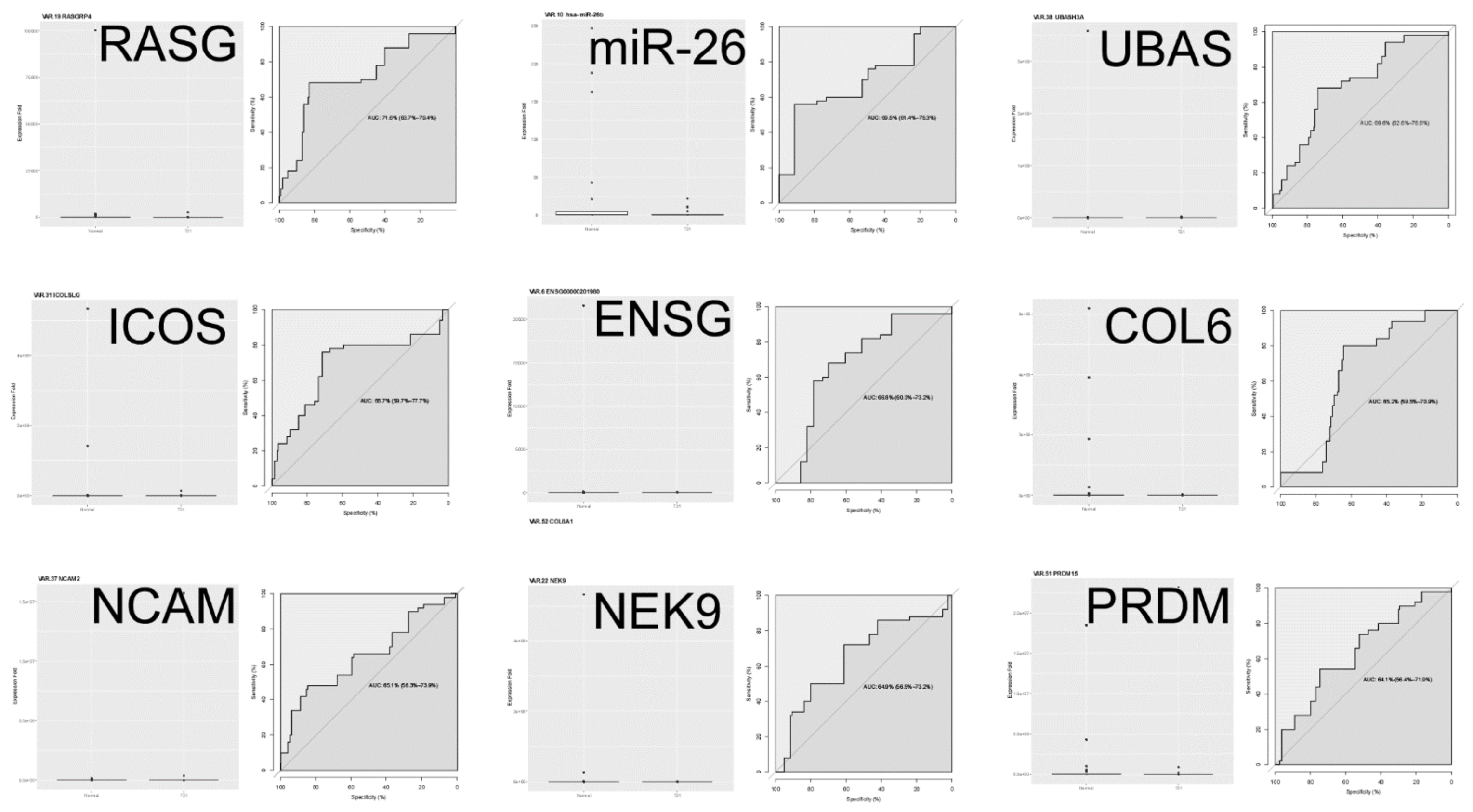
3.2.2. Validation Results
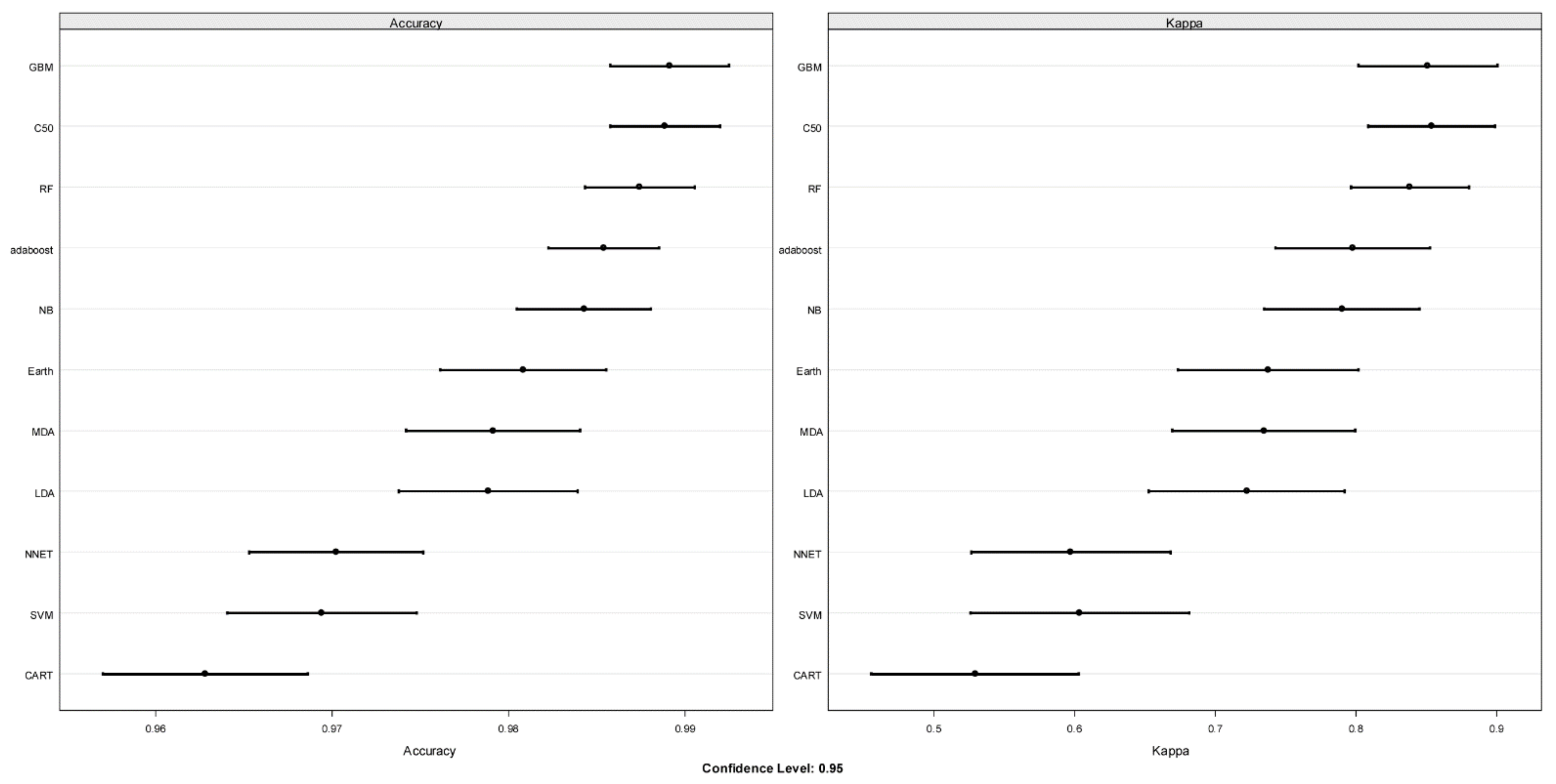
3.2.3. Application of ML Classification Algorithms to qRT-PCR Data
4. Discussion
4.1. Main Findings
4.2. Strengths and Limitations
4.3. ML Results
4.4. Interpretation
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schliep, K.C.; Feldkamp, M.L.; Hanson, H.A.; Hollingshaus, M.; Fraser, A.; Smith, K.R.; Panushka, K.A.; Varner, M.W. Are paternal or grandmaternal age associated with higher probability of trisomy 21 in offspring? A population-based, matched case-control study, 1995-2015. Paediatr. Perinat. Epidemiol. 2021, 35, 281–291. [Google Scholar] [CrossRef]
- Thompson, J.A. Disentangling the roles of maternal and paternal age on birth prevalence of down syndrome and other chromosomal disorders using a Bayesian modeling approach. BMC Med. Res. Methodol. 2019, 19, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S.; et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.T.; Isenburg, J.L.; Canfield, M.A.; Meyer, R.E.; Correa, A.; Alverson, C.J.; Lupo, P.J.; Riehle-Colarusso, T.; Cho, S.J.; Aggarwal, D.; et al. National population-based estimates for major birth defects, 2010-2014. Birth Defects Res. 2019, 111, 1420–1435. [Google Scholar] [CrossRef]
- de Groot-van der Mooren, M.D.; Tamminga, S.; Oepkes, D.; Weijerman, M.E.; Cornel, M.C. Older mothers and increased impact of prenatal screening: Stable livebirth prevalence of trisomy 21 in the Netherlands for the period 2000–2013. Eur. J. Hum. Genet. 2018, 26, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Potter, H.; Granic, A.; Caneus, J. Role of Trisomy 21 Mosaicism in sporadic and familial Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 7–17. [Google Scholar] [CrossRef]
- Schieve, L.A.; Boulet, S.L.; Kogan, M.D.; Van Naarden-Braun, K.; Boyle, C.A. A population-based assessment of the health, functional status, and consequent family impact among children with Down syndrome. Disabil. Health J. 2011, 4, 68–77. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, D.E.; Kim, S.Y.; Kim, H.J.; Kim, K.S.; Han, Y.J.; Kim, M.H.; Choi, J.S.; Kim, M.Y.; Ryu, H.M.; et al. MicroRNAs as potential biomarkers for noninvasive detection of fetal trisomy 21. J. Assist. Reprod Genet. 2015, 32, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Biggio, J.R., Jr.; Morris, T.C.; Owen, J.; Stringer, J.S. An outcomes analysis of five prenatal screening strategies for trisomy 21 in women younger than 35 years. Am. J. Obstet. Gynecol. 2004, 190, 721–729. [Google Scholar] [CrossRef]
- Chiu, R.W.; Chan, K.C.; Gao, Y.; Lau, V.Y.; Zheng, W.; Leung, T.Y.; Foo, C.H.; Xie, B.; Tsui, N.B.; Lun, F.M.; et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [Green Version]
- Guseh, S.H. Noninvasive prenatal testing: From aneuploidy to single genes. Hum. Genet. 2020, 139, 1141–1148. [Google Scholar] [CrossRef]
- Cuckle, H.S. Extending the provision of cell-free DNA screening for trisomy 21. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 447. [Google Scholar] [CrossRef]
- Hulstaert, F.; Neyt, M.; Gyselaers, W. The non-invasive prenatal test (NIPT) for trisomy 21—Health economic aspects. In Health Technology Assessment (HTA); Belgian Health Care Knowledge Centre (KCE): Brussels, Belgium, 2014. [Google Scholar]
- Huang, T.; Meschino, W.S.; Teitelbaum, M.; Dougan, S.; Okun, N. Enhanced First Trimester Screening for Trisomy 21 with Contingent Cell-Free Fetal DNA: A Comparative Performance and Cost Analysis. J. Obstet Gynaecol Can. 2017, 39, 742–749. [Google Scholar] [CrossRef]
- Devaux, Y. Transcriptome of blood cells as a reservoir of cardiovascular biomarkers. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.L.; Rood, I.M.; Deegens, J.K.J.; Klein, J.B. Isolation and characterization of urinary extracellular vesicles: Implications for biomarker discovery. Nat. Rev. Nephrol. 2017, 13, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Vea, A.; Llorente-Cortes, V.; de Gonzalo-Calvo, D. Circular RNAs in blood. Adv. Exp. Med. Biol. 2018, 1087, 119–130. [Google Scholar] [CrossRef]
- Adam, L.; Wszolek, M.F.; Liu, C.G.; Jing, W.; Diao, L.; Zien, A.; Zhang, J.D.; Jackson, D.; Dinney, C.P. Plasma microRNA profiles for bladder cancer detection. Urol. Oncol. 2013, 31, 1701–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulouarn, C.; Lefebvre, G.; Derambure, C.; Lequerre, T.; Scotte, M.; Francois, A.; Cellier, D.; Daveau, M.; Salier, J.P. Altered gene expression in acute systemic inflammation detected by complete coverage of the human liver transcriptome. Hepatology 2004, 39, 353–364. [Google Scholar] [CrossRef]
- Weiner, C.P.; Mason, C.W.; Dong, Y.; Buhimschi, I.A.; Swaan, P.W.; Buhimschi, C.S. Human effector/initiator gene sets that regulate myometrial contractility during term and preterm labor. Am. J. Obstet. Gynecol. 2010, 202, 474.e471–420. [Google Scholar] [CrossRef] [Green Version]
- Poon, L.L.; Leung, T.N.; Lau, T.K.; Lo, Y.M. Presence of fetal RNA in maternal plasma. Clin. Chem. 2000, 46, 1832–1834. [Google Scholar] [CrossRef] [Green Version]
- Heng, Y.J.; Pennell, C.E.; McDonald, S.W.; Vinturache, A.E.; Xu, J.; Lee, M.W.; Briollais, L.; Lyon, A.W.; Slater, D.M.; Bocking, A.D.; et al. Maternal whole blood gene expression at 18 and 28 weeks of gestation associated with spontaneous preterm birth in asymptomatic women. PLoS ONE 2016, 11, e0155191. [Google Scholar] [CrossRef] [Green Version]
- Luque, A.; Farwati, A.; Crovetto, F.; Crispi, F.; Figueras, F.; Gratacos, E.; Aran, J.M. Usefulness of circulating microRNAs for the prediction of early preeclampsia at first-trimester of pregnancy. Sci. Rep. 2014, 4, 4882. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Higashijima, A.; Miura, S.; Mishima, H.; Yamasaki, K.; Abe, S.; Hasegawa, Y.; Kaneuchi, M.; Yoshida, A.; Kinoshita, A.; et al. Predominantly placenta-expressed mRNAs in maternal plasma as predictive markers for twin-twin transfusion syndrome. Prenat. Diagn 2014, 34, 345–349. [Google Scholar] [CrossRef]
- Salomon, C.; Nuzhat, Z.; Dixon, C.L.; Menon, R. Placental exosomes during gestation: Liquid biopsies carrying signals for the regulation of human parturition. Curr. Pharm. Des. 2018, 24, 974–982. [Google Scholar] [CrossRef]
- Wander, P.L.; Boyko, E.J.; Hevner, K.; Parikh, V.J.; Tadesse, M.G.; Sorensen, T.K.; Williams, M.A.; Enquobahrie, D.A. Circulating early- and mid-pregnancy microRNAs and risk of gestational diabetes. Diabetes Res. Clin. Pract. 2017, 132, 1–9. [Google Scholar] [CrossRef]
- Whitehead, C.L.; Walker, S.P.; Tong, S. Measuring circulating placental RNAs to non-invasively assess the placental transcriptome and to predict pregnancy complications. Prenat. Diagn 2016, 36, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Yoffe, L.; Gilam, A.; Yaron, O.; Polsky, A.; Farberov, L.; Syngelaki, A.; Nicolaides, K.; Hod, M.; Shomron, N. Early detection of preeclampsia using circulating small non-coding RNA. Sci. Rep. 2018, 8, 3401. [Google Scholar] [CrossRef]
- Zhu, Y.; Tian, F.; Li, H.; Zhou, Y.; Lu, J.; Ge, Q. Profiling maternal plasma microRNA expression in early pregnancy to predict gestational diabetes mellitus. Int. J. Gynaecol. Obstet. 2015, 130, 49–53. [Google Scholar] [CrossRef]
- Giannopoulou, L.; Zavridou, M.; Kasimir-Bauer, S.; Lianidou, E.S. Liquid biopsy in ovarian cancer: The potential of circulating miRNAs and exosomes. Transl. Res. 2019, 205, 77–91. [Google Scholar] [CrossRef]
- Li, L.M.; Liu, H.; Liu, X.H.; Hu, H.B.; Liu, S.M. Clinical significance of exosomal miRNAs and proteins in three human cancers with high mortality in China. Oncol. Lett. 2019, 17, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Hille, C.; Scher, H.I. Circulating tumor cells in prostate cancer: From discovery to clinical utility. Clin. Chem. 2019, 65, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Ruhen, O.; Meehan, K. Tumor-derived extracellular vesicles as a novel source of protein biomarkers for cancer diagnosis and monitoring. Proteomics 2019, 19, e1800155. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, F.; Ding, H.; Wang, Y.; Li, P.; Wang, K. Emerging function and clinical values of exosomal MicroRNAs in cancer. Mol. Ther. Nucleic Acids 2019, 16, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Erturk, B.; Karaca, E.; Aykut, A.; Durmaz, B.; Guler, A.; Buke, B.; Yeniel, A.O.; Ergenoglu, A.M.; Ozkinay, F.; Ozeren, M.; et al. Prenatal evaluation of MicroRNA expressions in pregnancies with down syndrome. Biomed. Res. Int. 2016, 2016, 5312674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbucka-Kretowska, M.; Niemira, M.; Paczkowska-Abdulsalam, M.; Bielska, A.; Szalkowska, A.; Parfieniuk, E.; Ciborowski, M.; Wolczynski, S.; Kretowski, A. Prenatal circulating microRNA signatures of foetal Down syndrome. Sci. Rep. 2019, 9, 2394. [Google Scholar] [CrossRef] [Green Version]
- Kamhieh-Milz, J.; Moftah, R.F.; Bal, G.; Futschik, M.; Sterzer, V.; Khorramshahi, O.; Burow, M.; Thiel, G.; Stuke-Sontheimer, A.; Chaoui, R.; et al. Differentially expressed microRNAs in maternal plasma for the noninvasive prenatal diagnosis of Down syndrome (trisomy 21). Biomed. Res. Int. 2014, 2014, 402475. [Google Scholar] [CrossRef] [PubMed]
- Kotlabova, K.; Doucha, J.; Chudoba, D.; Calda, P.; Dlouha, K.; Hromadnikova, I. Extracellular chromosome 21-derived microRNAs in euploid & aneuploid pregnancies. Indian J. Med. Res. 2013, 138, 935–943. [Google Scholar] [PubMed]
- Svobodova, I.; Korabecna, M.; Calda, P.; Brestak, M.; Pazourkova, E.; Pospisilova, S.; Krkavcova, M.; Novotna, M.; Horinek, A. Differentially expressed miRNAs in trisomy 21 placentas. Prenat. Diagn 2016, 36, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Zednikova, I.; Chylikova, B.; Seda, O.; Korabecna, M.; Pazourkova, E.; Brestak, M.; Krkavcova, M.; Calda, P.; Horinek, A. Genome-wide miRNA profiling in plasma of pregnant women with down syndrome fetuses. Mol. Biol. Rep. 2020, 47, 4531–4540. [Google Scholar] [CrossRef] [PubMed]
- Weiner, C.P.; Dong, Y.; Zhou, H.; Cuckle, H.; Ramsey, R.; Egerman, R.; Buhimschi, I.; Buhimschi, C. Early pregnancy prediction of spontaneous preterm birth before 32 completed weeks of pregnancy using plasma RNA: Transcriptome discovery and initial validation of an RNA panel of markers. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.Y.; Gao, F.J.; Xu, P.; Zhang, S.H.; Wu, J.H. Cell development deficiency and gene expression dysregulation of trisomy 21 retina revealed by single-nucleus RNA sequencing. Front. Bioeng. Biotechnol. 2020, 8, 564057. [Google Scholar] [CrossRef]
- Laurent, A.P.; Siret, A.; Ignacimouttou, C.; Panchal, K.; Diop, M.; Jenni, S.; Tsai, Y.C.; Roos-Weil, D.; Aid, Z.; Prade, N.; et al. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in down syndrome B-cell Leukemia. Clin. Cancer Res. 2020, 26, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Megarbane, A.; Piquemal, D.; Rebillat, A.S.; Stora, S.; Pierrat, F.; Bruno, R.; Noguier, F.; Mircher, C.; Ravel, A.; Vilaire-Meunier, M.; et al. Transcriptomic study in women with trisomy 21 identifies a possible role of the GTPases of the immunity-associated proteins (GIMAP) in the protection of breast cancer. Sci. Rep. 2020, 10, 9447. [Google Scholar] [CrossRef]
- Ponroy Bally, B.; Farmer, W.T.; Jones, E.V.; Jessa, S.; Kacerovsky, J.B.; Mayran, A.; Peng, H.; Lefebvre, J.L.; Drouin, J.; Hayer, A.; et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 2020, 29, 785–802. [Google Scholar] [CrossRef]
- Stamoulis, G.; Garieri, M.; Makrythanasis, P.; Letourneau, A.; Guipponi, M.; Panousis, N.; Sloan-Bena, F.; Falconnet, E.; Ribaux, P.; Borel, C.; et al. Single cell transcriptome in aneuploidies reveals mechanisms of gene dosage imbalance. Nat. Commun. 2019, 10, 4495. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Han, Y.J.; Kim, H.J.; Kim, M.Y.; Park, S.Y.; Cho, Y.H.; Ryu, H.M. Integrative analyses of genes and microRNA expressions in human trisomy 21 placentas. BMC Med. Genom. 2018, 11, 46. [Google Scholar] [CrossRef] [Green Version]
- Xing, K.; Cui, Y.; Luan, J.; Zhou, X.; Shi, L.; Han, J. Establishment of a human trisomy 18 induced pluripotent stem cell line from amniotic fluid cells. Intractable Rare Dis. Res. 2018, 7, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volk, M.; Maver, A.; Hodzic, A.; Lovrecic, L.; Peterlin, B. Transcriptome profiling uncovers potential common mechanisms in fetal trisomies 18 and 21. Omics J. Integr. Biol. 2017, 21, 565–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Ribas, I.; Diaz-Gimeno, P.; Sebastian-Leon, P.; Mercader, A.; Quinonero, A.; Ballesteros, A.; Pellicer, A.; Dominguez, F. Transcriptomic behavior of genes associated with chromosome 21 aneuploidies in early embryo development. Fertil. Steril. 2019, 111, 991–1001 e1002. [Google Scholar] [CrossRef]
- Yang, J.; Ding, X.; Zhu, W. Improving the calling of non-invasive prenatal testing on 13-/18-/21-trisomy by support vector machine discrimination. PLoS ONE 2018, 13, e0207840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J. Minimum standards set out for gene-expression data. Nature 2002, 415, 946. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef] [Green Version]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Lunardon, N.; Menardi, G.; Torelli, N. ROSE: A package for binary imbalanced learning. R J. 2014, 6, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Hockberg, Y. A sharper Bonferroni precedure for multiple tests of significance. Biometrika 1988, 75, 800–802. [Google Scholar] [CrossRef]
- Hommel, G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biomerika 1988, 75, 800–803. [Google Scholar] [CrossRef]
- Bonferroni, C.E. Teoria statistica delle classi e calcolo delle probabilità. In Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze; Seeber: Johannesburg, South Africa, 1936. [Google Scholar]
- Alldred, S.K.; Takwoingi, Y.; Guo, B.; Pennant, M.; Deeks, J.J.; Neilson, J.P.; Alfirevic, Z. First and second trimester serum tests with and without first trimester ultrasound tests for Down’s syndrome screening. Cochrane Database Syst. Rev. 2017, 3, CD012599. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.S.; Ishibashi, O.; Ishikawa, G.; Ishikawa, T.; Katayama, A.; Mishima, T.; Takizawa, T.; Shigihara, T.; Goto, T.; Izumi, A.; et al. Human villous trophoblasts express and secrete placenta-specific microRNAs into maternal circulation via exosomes. Biol. Reprod. 2009, 81, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Hadlich, F.; Abbas, M.W.; Iqbal, M.A.; Tesfaye, D.; Bouma, G.J.; Winger, Q.A.; Ponsuksili, S. MicroRNA-mRNA networks in pregnancy complications: A comprehensive downstream analysis of potential biomarkers. Int. J. Mol. Sci. 2021, 22, 2313. [Google Scholar] [CrossRef] [PubMed]
- Munchel, S.; Rohrback, S.; Randise-Hinchliff, C.; Kinnings, S.; Deshmukh, S.; Alla, N.; Tan, C.; Kia, A.; Greene, G.; Leety, L.; et al. Circulating transcripts in maternal blood reflect a molecular signature of early-onset preeclampsia. Sci. Transl. Med. 2020, 12, eaaz0131. [Google Scholar] [CrossRef]
- Filant, J.; Nejad, P.; Paul, A.; Simonson, B.; Srinivasan, S.; Zhang, X.; Balaj, L.; Das, S.; Gandhi, R.; Laurent, L.C.; et al. Isolation of extracellular RNA from Serum/Plasma. Methods Mol. Biol. 2018, 1740, 43–57. [Google Scholar] [CrossRef]
- Li, X.; Mauro, M.; Williams, Z. Comparison of plasma extracellular RNA isolation kits reveals kit-dependent biases. Biotechniques 2015, 59, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papavassiliou, P.; York, T.P.; Gursoy, N.; Hill, G.; Nicely, L.V.; Sundaram, U.; McClain, A.; Aggen, S.H.; Eaves, L.; Riley, B.; et al. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am. J. Med. Genet. A 2009, 149A, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Sparks, T.N.; Thao, K.; Norton, M.E. Mosaic trisomy 16: What are the obstetric and long-term childhood outcomes? Genet. Med. 2017, 19, 1164–1170. [Google Scholar] [CrossRef]
- Batuwita, R.; Palade, V. Class imbalance learning methods for support vector machines. In Imbalanced Learning: Foundations, Algorithms, and Applications; He, H., Ma, Y., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Buda, M.; Maki, A.; Mazurowski, M.A. A systematic study of the class imbalance problem in convolutional neural networks. Neural Netw. 2018, 106, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Yin, Y.; Dong, C.; Yang, G.; Zhou, G. On the class imbalance problem. In Proceedings of the Fourth International Conference on Natural Compuation, Jinan, China, 18–20 October 2008; IEEE Computer Society: Washington, DC, USA, 2008. [Google Scholar]
- Wang, Y.C.; Cheng, C.H. A multiple combined method for rebalancing medical data with class imbalances. Comput. Biol. Med. 2021, 134, 104527. [Google Scholar] [CrossRef]
- Niculescu, A.B.; Le-Niculescu, H. Precision medicine in psychiatry: Biomarkers to the forefront. Neuropsychopharmacology 2022, 47, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.B.; Le-Niculescu, H. Convergence of recent GWAS data for suicidality with previous blood biomarkers: Independent reproducibility using independent methodologies in independent cohorts. Mol. Psychiatry 2020, 25, 19–21. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T21 (n = 10) Mean (SD) | Range | Normal (n = 10) Mean (SD) | Range | T21 p-Value * | Race and Ethnicity p-Value * | |
|---|---|---|---|---|---|---|
| GA (week) | 12.9 (0.6) | (11.9–13.9) | 12.7 (0.5) | (12.1–13.4) | NS | NS |
| MA (years) | 37.3 (4.1) | (27.2–42.0) | 33.7 (5.0) | (24.6–40.0) | p = 0.098 ** | NS *** |
| Height (cm) | 163.6 (4.9) | (157.5–170.2) | 165.1 (4.4) | (157.5–171.0) | NS | NS |
| Weight (kg) | 70.7 (9.9) | (60.0–83.5) | 69.8 (18.7) | (50.00–115.0) | NS *** | NS *** |
| (a) | ||||
|---|---|---|---|---|
| Group 1. Coding mRNA Gene Name | p-Value | Ref Sequence | Chromosome Origin | Up or Down Regulation |
| SORBS2-Hs00243432_m1 | <0.01 | NM_001145671 | 4 | Down |
| SORBS2-Hs01125202_m1 | <0.01 | NM_001145671 | 4 | Up |
| DSCAM-Hs00242097_m1 | <0.01 | NM_020693 | 11 | Down |
| NEK9-Hs00929602_m1 | <0.01 | NM_033116 | 14 | Down |
| NEK9-Hs00929594_m1 | <0.01 | NM_033116 | 14 | Up |
| ABCC1-Hs01561504_m1 | <0.01 | NM_004996 | 16 | Up |
| FAM20A-Hs01034071_m1 | <0.01 | NR_027751 | 17 | Down |
| FAM20A-Hs01034070_m1 | <0.01 | NR_027751 | 17 | Down |
| RASGRP4-Hs01073179_m1 | <0.01 | NM_170604 | 19 | Down |
| TMPRSS2-ERG fusion gene | <0.01 | NM_002772 | 21 | Down |
| ATP5O-Hs04272738_m1 | <0.01 | NM_001697.3 | 21 | Down |
| ICOSLG-Hs00391287_m1 | <0.01 | NM_015259 | 21 | Down |
| DOP1B-Hs01123288_m1 | <0.01 | NM_005128 | 21 | Down |
| DOP1B-Hs01123267_g1 | <0.01 | NM_005128 | 21 | Down |
| C21orf33-Hs01105802_g1 | <0.01 | NM_004649 | 21 | Down |
| ADAMTS5-Hs04272736_s1 | <0.01 | NM_007038 | 21 | Down |
| CXADR-Hs04194411_s1 | <0.01 | NM_001338 | 21 | Down |
| UBASH3A-Hs00955168_m1 | <0.01 | NM_001001895.3 | 21 | Down |
| CHODL-Hs01070471_m1 | <0.01 | NM_024944.3 | 21 | Down |
| PKNOX1-Hs01007098_m1 | <0.01 | NM_004571 | 21 | Down |
| PKNOX1-Hs01007097_m1 | <0.01 | NM_001286258 | 21 | Down |
| PKNOX1-Hs00231814_m1 | <0.01 | 21 | Down | |
| SLC19A1-Hs00953341_m1 | <0.01 | NM_194255 | 21 | Down |
| PRDM15-Hs00411318_m1 | <0.01 | NM_022115 | 21 | Down |
| COL6A1-Hs01095585_m1 | <0.01 | NM_001848 | 21 | Down |
| ABCG1-Hs01555191_m1 | <0.01 | NM_016818 | 21 | Down |
| GART-Hs00531926_m1 | <0.01 | NM_000819 | 21 | Down |
| ERG-Hs01573964_m1 | <0.01 | NM_004449 | 21 | Up |
| NCAM2-Hs01562292_m1 | <0.01 | NM_004540.5 | 21 | Up |
| UBASH3A-Hs00955169_m1 | <0.01 | NM_018961.4 | 21 | Up |
| PFKL-Hs01040525_m1 | <0.01 | NR_024108 | 21 | Up |
| PKNOX1-Hs01007094_m1 | <0.01 | NM_001320694 | 21 | Up |
| PKNOX1-Hs01007093_m1 | <0.01 | 21 | Up | |
| PKNOX1-Hs01007092_m1 | <0.01 | NM_004571 | 21 | Up |
| CYYR1-Hs00951849_m1 | <0.01 | NR_135472 | 21 | Up |
| SLC19A1-Hs00953342_m1 | <0.01 | NM_194255 | 21 | Up |
| (b) | ||||
| Group 2. Noncoding Small RNA Gene Name | p-Value | Type | Chromosome Origin | Up or Down Regulation |
| hsa-mir-26b | <0.01 | miRNA | 2 | Down |
| hsa-mir-216b | <0.01 | miRNA | 2 | Up |
| hsa-mir-569 F1 | <0.01 | miRNA | 3 | Down |
| hsa-mir-548I | <0.01 | miRNA | 3 | Down |
| ENSG00000212363 | <0.01 | snoRNA | 5 | Down |
| hsa-mir-581 F1 | <0.01 | miRNA | 5 | Up |
| HBII-276 F2 | <0.01 | CDBox | 8 | Up |
| hsa-let-7d F1 | <0.01 | miRNA | 9 | Up |
| ENSG00000201980 | <0.01 | snoRNA | 11 | Up |
| ENSG00000199282 | <0.01 | snoRNA | 13 | Down |
| hsa-mir-376a-2/1 F2 | <0.01 | miRNA | 14 | Down |
| ENSG00000199633 F2 | <0.01 | snoRNA | 15 | Up |
| ENSG00000199856 F1 | <0.01 | snoRNA | 18 | Down |
| hsa-mir-523 | <0.01 | miRNA | 19 | Down |
| ENSG00000207147 F2 | <0.01 | snoRNA | 21 | Up |
| ENSG00000202231 | <0.01 | snoRNA | X | Down |
| hsa-mir-98 | <0.01 | miRNA | X | Down |
| hsa-mir-450b | <0.01 | miRNA | X | Down |
| T21 (n = 50) Mean (SD) | Range | Normal (n = 948) Mean (SD) | Range | T21 p-Value * | Race/Ethnicity p-Value * | |
|---|---|---|---|---|---|---|
| GA (week) | 13.0 (0.7) | (11.3–14.1) | 12.7 (0.6) | (11.2–14.1) | <0.001 | NS |
| MA (years) | 37.6 (4.4) | (26.4–46) | 31.7 (5.6) | (18.1–45.1) | <0.001 | <0.001 |
| Height (cm) | 164.6 (7.1) | (149.9–182.9) | 164.5 (6.9) | (138.0–195.6) | NS | <0.001 |
| Weight (kg) | 68.1 (11.1) | (44.5–99.2) | 66.6 (11.9) | (40.0–29.0) | NS | <0.001 |
| Chromosome | Plate Position | Variables | NCBI Names | Mann–Whitney–Wilcoxon | Q-Values | Benjamini–Hochberg | Benjamini–Yekutieli | Holm | Hochberg | Hommel | Bonferroni |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MA | 1.64 × 10−12 | 8.34 × 10−11 | 9.51 × 10−11 | 4.42 × 10−10 | 9.51 × 10−11 | 9.51 × 10−11 | 9.51 × 10−11 | 9.51 × 10−11 | |||
| 19 | 19 | RASGRP4 | NM_170604 | 2.49 × 10−7 | 6.33 × 10−6 | 7.21 × 10−6 | 3.35 × 10−5 | 1.42 × 10−5 | 1.42 × 10−5 | 1.42 × 10−5 | 1.44 × 10−5 |
| 2 | 10 | hsa-mir-26b | miRNA miR-26b | 2.16 × 10−6 | 3.52 × 10−5 | 4.01 × 10−5 | 1.86 × 10−4 | 1.21 × 10−4 | 1.21 × 10−4 | 1.19 × 10−4 | 1.25 × 10−4 |
| 21 | 38 | UBASH3A | NM_018961.4 | 2.77 × 10−6 | 3.52 × 10−5 | 4.01 × 10−5 | 1.86 × 10−4 | 1.52 × 10−4 | 1.52 × 10−4 | 1.52 × 10−4 | 1.60 × 10−4 |
| 21 | 31 | ICOSLG | NM_015259 | 7.77 × 10−6 | 7.91 × 10−5 | 9.01 × 10−5 | 4.19 × 10−4 | 4.20 × 10−4 | 4.20 × 10−4 | 4.20 × 10−4 | 4.51 × 10−4 |
| 11 | 9 | ENSG00000201980 | snoRNA SNORA62L4 | 6.24 × 10−5 | 5.29 × 10−4 | 6.03 × 10−4 | 2.80 × 10−3 | 3.31 × 10−3 | 3.31 × 10−3 | 3.31 × 10−3 | 3.62 × 10−3 |
| GA.w | 2.51 × 10−4 | 1.68 × 10−3 | 1.92 × 10−3 | 8.92 × 10−3 | 1.30 × 10−2 | 1.30 × 10−2 | 1.23 × 10−2 | 1.45 × 10−2 | |||
| 21 | 52 | COL6A1 | NM_001848 | 2.85 × 10−4 | 1.68 × 10−3 | 1.92 × 10−3 | 8.92 × 10−3 | 1.45 × 10−2 | 1.45 × 10−2 | 1.40 × 10−2 | 1.65 × 10−2 |
| 21 | 37 | NCAM2 | NM_004540.5 | 2.98 × 10−4 | 1.68 × 10−3 | 1.92 × 10−3 | 8.92 × 10−3 | 1.49 × 10−2 | 1.49 × 10−2 | 1.46 × 10−2 | 1.73 × 10−2 |
| 14 | 22 | NEK9 | NM_033116 | 3.78 × 10−4 | 1.92 × 10−3 | 2.19 × 10−3 | 1.02 × 10−2 | 1.85 × 10−2 | 1.85 × 10−2 | 1.81 × 10−2 | 2.19 × 10−2 |
| 21 | 51 | PRDM15 | NM_022115 | 7.18 × 10−4 | 3.32 × 10−3 | 3.78 × 10−3 | 1.76 × 10−2 | 3.44 × 10−2 | 3.44 × 10−2 | 3.30 × 10−2 | 4.16 × 10−2 |
| 21 | 2 | ENSG00000207147 F2 | snoRNA SNORA51L12 | 1.41 × 10−3 | 5.96 × 10−3 | 6.80 × 10−3 | 3.16 × 10−2 | 6.64 × 10−2 | 6.64 × 10−2 | 6.32 × 10−2 | 8.19 × 10−2 |
| 21 | 27 | TMPRSS2-ERG fusion gene | NM_002772 | 1.52 × 10−3 | 5.96 × 10−3 | 6.80 × 10−3 | 3.16 × 10−2 | 7.01 × 10−2 | 7.01 × 10−2 | 6.70 × 10−2 | 8.84 × 10−2 |
| 18 | 17 | ENSG00000199856 F1 | snoRNA SNODB852 | 2.03 × 10−3 | 7.39 × 10−3 | 8.42 × 10−3 | 3.91 × 10−2 | 9.15 × 10−2 | 9.15 × 10−2 | 8.95 × 10−2 | 1.18 × 10−1 |
| 21 | 33 | DOP1B | NM_005128 | 3.08 × 10−3 | 1.04 × 10−2 | 1.19 × 10−2 | 5.53 × 10−2 | 1.36 × 10−1 | 1.36 × 10−1 | 1.32 × 10−1 | 1.79 × 10−1 |
| 21 | 26 | SORBS2 | NM_001145671 | 4.21 × 10−3 | 1.34 × 10−2 | 1.53 × 10−2 | 7.10 × 10−2 | 1.81 × 10−1 | 1.81 × 10−1 | 1.81 × 10−1 | 2.44 × 10−1 |
| 9 | 3 | hsa-let-7d F1 | miRNA let-7d | 1.08 × 10−2 | 3.22 × 10−2 | 3.67 × 10−2 | 1.70 × 10−1 | 4.52 × 10−1 | 4.52 × 10−1 | 4.19 × 10−1 | 6.24 × 10−1 |
| 17 | 20 | FAM20A | NR_027751 | 1.92 × 10−2 | 5.20 × 10−2 | 5.93 × 10−2 | 2.76 × 10−1 | 7.88 × 10−1 | 7.77 × 10−1 | 6.15 × 10−1 | 1.00 × 100 |
| 21 | 41 | CHODL | NM_024944.3 | 1.94 × 10−2 | 5.20 × 10−2 | 5.93 × 10−2 | 2.76 × 10−1 | 7.88 × 10−1 | 7.77 × 10−1 | 6.22 × 10−1 | 1.00 × 100 |
| X | 12 | hsa-mir-450b | miRNA miR-450b | 2.79 × 10−2 | 7.10 × 10−2 | 8.10 × 10−2 | 3.76 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 7.71 × 10−1 | 1.00 × 100 |
| 17 | 21 | FAM20A | NR_027751 | 3.41 × 10−2 | 8.25 × 10−2 | 9.41 × 10−2 | 4.37 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 8.51 × 10−1 | 1.00 × 100 |
| 21 | 34 | C21orf33 | NM_004649 | 5.11 × 10−2 | 1.18 × 10−1 | 1.35 × 10−1 | 6.26 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 11 | 29 | DSCAM | NM_020693 | 5.46 × 10−2 | 1.21 × 10−1 | 1.38 × 10−1 | 6.40 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 36 | CXADR | NM_001338 | 6.04 × 10−2 | 1.28 × 10−1 | 1.46 × 10−1 | 6.79 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 13 | 14 | ENSG00000199282 | snoRNA SNOFA9 | 6.63 × 10−2 | 1.35 × 10−1 | 1.54 × 10−1 | 7.14 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 30 | ERG | NM_004449 | 8.44 × 10−2 | 1.65 × 10−1 | 1.88 × 10−1 | 8.75 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 3 | 4 | hsa-mir-569 F1 | miRNA miR-569 | 9.42 × 10−2 | 1.77 × 10−1 | 2.02 × 10−1 | 9.40 × 10−1 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 5 | 13 | SORBS2 | NM_001145671 | 1.09 × 10−1 | 1.91 × 10−1 | 2.18 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 4 | 25 | ENSG00000212363 | snoRNA SNOFA40L2 | 1.09 × 10−1 | 1.91 × 10−1 | 2.18 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 47 | PKNOX1 | 1.20 × 10−1 | 2.04 × 10−1 | 2.33 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | |
| 2 | 8 | hsa-mir-216b | miRNA miR-216b | 1.52 × 10−1 | 2.50 × 10−1 | 2.85 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 32 | DOP1B | NM_005128 | 1.63 × 10−1 | 2.59 × 10−1 | 2.95 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 14 | 16 | hsa-mir-376a-2/1 F2 | miRNA miR-376a | 1.86 × 10−1 | 2.86 × 10−1 | 3.26 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 35 | ADAMTS5 | NM_007038 | 1.91 × 10−1 | 2.86 × 10−1 | 3.26 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| Weight | 2.08 × 10−1 | 3.02 × 10−1 | 3.44 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | |||
| 5 | 11 | hsa-mir-581 F1 | miRNA miR-581 | 2.20 × 10−1 | 3.11 × 10−1 | 3.55 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 46 | PKNOX1 | NM_004571 | 2.37 × 10−1 | 3.20 × 10−1 | 3.64 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 44 | PKNOX1 | NM_001320694 | 2.39 × 10−1 | 3.20 × 10−1 | 3.64 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 50 | SLC19A1 | NM_194255 | 2.97 × 10−1 | 3.81 × 10−1 | 4.34 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 40 | PFKL | NR_024108 | 2.99 × 10−1 | 3.81 × 10−1 | 4.34 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 8 | 18 | HBII-276 F2 | SnoRNA HBII-276 CDBox | 3.33 × 10−1 | 4.13 × 10−1 | 4.71 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 15 | 1 | ENSG00000199633 F2 | snoRNA SNODB1383 | 3.62 × 10−1 | 4.38 × 10−1 | 5.00 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| X | 7 | ENSG00000202231 | snoRNA SNOFA9 | 3.74 × 10−1 | 4.43 × 10−1 | 5.05 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 28 | ATP5O | NM_001697.3 | 3.85 × 10−1 | 4.46 × 10−1 | 5.08 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 49 | SLC19A1 | NM_194255 | 4.72 × 10−1 | 5.33 × 10−1 | 6.08 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 4 | 24 | ABCC1 | NM_004996 | 5.60 × 10−1 | 6.20 × 10−1 | 7.06 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 3 | 5 | hsa-mir-548I | miRNA miR-548I | 6.62 × 10−1 | 7.12 × 10−1 | 8.12 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 45 | PKNOX1 | 6.93 × 10−1 | 7.12 × 10−1 | 8.12 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | |
| 21 | 48 | CYYR1 | NR_135472 | 6.99 × 10−1 | 7.12 × 10−1 | 8.12 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| X | 9 | hsa-mir-98 | miRNA miR-98 | 7.00 × 10−1 | 7.12 × 10−1 | 8.12 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 42 | PKNOX1 | NM_004571 | 7.30 × 10−1 | 7.28 × 10−1 | 8.30 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 54 | GART | NM_000819 | 7.53 × 10−1 | 7.37 × 10−1 | 8.40 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 14 | 23 | NEK9 | NM_033116 | 8.48 × 10−1 | 8.14 × 10−1 | 9.28 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 19 | 15 | hsa-mir-523 | miRNA miR-523 | 8.96 × 10−1 | 8.44 × 10−1 | 9.62 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| Height | 9.63 × 10−1 | 8.75 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | |||
| 21 | 43 | PKNOX1 | NM_001286258 | 9.72 × 10−1 | 8.75 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 39 | UBASH3A | NM_001001895.3 | 9.93 × 10−1 | 8.75 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| 21 | 53 | ABCG1 | NM_016818 | 9.98 × 10−1 | 8.75 × 10−1 | 9.98 × 10−1 | 1.00 × 100 | 1.00 × 100 | 9.98 × 10−1 | 9.98 × 10−1 | 1.00 × 100 |
| GBM | 70% Training | 75% Training | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Up sample | Accuracy | 1.000 | Kappa | 1.000 | Up sample | Accuracy | 0.992 | Kappa | 0.912 |
| Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight | Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight |
| RASGRP4 | NM_170604 | 19 | 166.77 | 0.193 | hsa-mir-26b | miRNA | 2 | 179.19 | 0.196 |
| hsa-mir-26b | miRNA | 2 | 131.32 | 0.152 | RASGRP4 | NM_170604 | 19 | 121.42 | 0.133 |
| UBASH3A | NM_018961.4 | 21 | 91.68 | 0.106 | MA | 85.02 | 0.093 | ||
| NCAM2 | NM_004540.5 | 21 | 88.98 | 0.103 | NCAM2 | NM_004540.5 | 21 | 82.56 | 0.090 |
| COL6A1 | NM_001848 | 21 | 56.96 | 0.066 | ENSG00000207147 F2 | snoRNA | 21 | 76.16 | 0.083 |
| MA | 51.68 | 0.060 | UBASH3A | NM_018961.4 | 21 | 71.55 | 0.078 | ||
| ICOSLG | NM_015259 | 21 | 50.45 | 0.058 | ENSG00000199856 F1 | snoRNA | 18 | 52.92 | 0.058 |
| C5.0 | 80% training | 70% training | |||||||
| Original | Accuracy | 1.000 | Kappa | 1.000 | Up sample | Accuracy | 0.99 | Kappa | 0.8837 |
| Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight | Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight |
| GART | mRNA/NM_000819 | 21 | 100.00 | 0.052 | hsa-mir-98 | miRNA | X | 70.78 | 0.260 |
| hsa-mir-26b | miRNA | 2 | 100.00 | 0.052 | hsa-mir-523 | miRNA | 19 | 69.05 | 0.253 |
| hsa-mir-450b | miRNA | X | 99.50 | 0.052 | ENSG00000207147 F2 | snoRNA | 21 | 60.02 | 0.220 |
| COL6A1 | NM_001848 | 21 | 98.87 | 0.052 | hsa-mir-569 F1 | miRNA | 3 | 50.98 | 0.187 |
| ATP5O | NM_001697.3 | 21 | 98.62 | 0.051 | hsa-mir-216b | miRNA | 2 | 21.84 | 0.080 |
| ENSG00000199633 F2 | snoRNA | 15 | 98.25 | 0.051 | |||||
| DOP1B | NM_005128 | 21 | 98.12 | 0.051 | |||||
| RF | 80% training | 80% training | |||||||
| Up sample | Accuracy | 0.995 | Kappa | 0.945 | Original | Accuracy | 0.995 | Kappa | 0.945 |
| Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight | Gene Name | Ref Sequence | Chromosome | Attribute usage | Weight |
| hsa-mir-26b | miRNA | 2 | 31.29 | 0.085 | GART | mRNA/NM_000819 | 21 | 4.01 | 0.105 |
| RASGRP4 | NM_170604 | 19 | 27.15 | 0.073 | hsa-mir-26b | miRNA | 2 | 2.73 | 0.071 |
| MA | 25.19 | 0.068 | hsa-mir-450b | miRNA | X | 2.61 | 0.068 | ||
| ICOSLG | NM_015259 | 21 | 20.69 | 0.056 | PKNOX1 | NM_004571 | 21 | 2.15 | 0.056 |
| UBASH3A | NM_018961.4 | 21 | 20.02 | 0.054 | ENSG00000199282 | snoRNA | 13 | 2.10 | 0.055 |
| DOP1B | NM_005128 | 21 | 19.74 | 0.053 | hsa-mir-376a-2/1 F2 | miRNA | 14 | 2.03 | 0.053 |
| FAM20A | NR_027751 | 17 | 17.87 | 0.048 | ATP5O | NM_001697.3 | 21 | 1.89 | 0.049 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiner, C.P.; Weiss, M.L.; Zhou, H.; Syngelaki, A.; Nicolaides, K.H.; Dong, Y. Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA. Diagnostics 2022, 12, 1410. https://doi.org/10.3390/diagnostics12061410
Weiner CP, Weiss ML, Zhou H, Syngelaki A, Nicolaides KH, Dong Y. Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA. Diagnostics. 2022; 12(6):1410. https://doi.org/10.3390/diagnostics12061410
Chicago/Turabian StyleWeiner, Carl P., Mark L. Weiss, Helen Zhou, Argyro Syngelaki, Kypros H. Nicolaides, and Yafeng Dong. 2022. "Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA" Diagnostics 12, no. 6: 1410. https://doi.org/10.3390/diagnostics12061410
APA StyleWeiner, C. P., Weiss, M. L., Zhou, H., Syngelaki, A., Nicolaides, K. H., & Dong, Y. (2022). Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA. Diagnostics, 12(6), 1410. https://doi.org/10.3390/diagnostics12061410