
When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms

Abstract
:1. Introduction
2. Diagnosis
3. The Different Forms of Sideroblastic Anemia
3.1. Congenital Sideroblastic Anemias
3.1.1. Non-Syndromic Forms
3.1.2. Syndromic Forms
3.2. Secondary Acquired Sideroblastic Anemias
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mc, F.A.; Davis, L.J. Iron-staining erythrocytic inclusions with especial reference to acquired haemolytic anaemia. Glasg. Med. J. 1947, 28, 237–279. [Google Scholar]
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellstrom-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717. [Google Scholar] [CrossRef]
- Cazzola, M.; Invernizzi, R. Ring sideroblasts and sideroblastic anemias. Haematologica 2011, 96, 789–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzola, M. Ineffective erythropoiesis and its treatment. Blood 2022, 139, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Introduction to a How I Treat series on acquired hemolytic anemia. Blood 2021, 137, 1269. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO classification of tumors of haematopoeitic and lymphoid tissues. In WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; IARC: Lyon, France, 2017; pp. 98–106. [Google Scholar]
- Long, B.; Shi, H.; Zhu, C. Clinical analysis and literature review of a case with the myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Hematology 2020, 25, 283–285. [Google Scholar] [CrossRef]
- Nathan, D.I.; Feld, J.; El Jamal, S.M.; Mascarenhas, J.; Tremblay, D. Myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis: Ringing in a new future. Leuk. Res. 2022, 115, 106820. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Steensma, D.P. Dysplasia has A differential diagnosis: Distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr. Hematol. Malig. Rep. 2012, 7, 310–320. [Google Scholar] [CrossRef]
- Solis, M.; Perrin, J.; Guedenet, J.C.; Lesesve, J.F. RBCs inclusions after splenectomy: Not only Howell-Jolly bodies! Ann. Biol. Clin. 2013, 71, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, G. Transient appearance of ring sideroblasts in peripheral blood in the acute phase of secondary hemolytic anemia. Blood 2019, 133, 1000. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J. Clin. Investig. 2020, 130, 5245–5256. [Google Scholar] [CrossRef]
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Long, Z.; Li, H.; Du, Y.; Han, B. Congenital sideroblastic anemia: Advances in gene mutations and pathophysiology. Gene 2018, 668, 182–189. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Richardson, D.R.; Prchal, J.; Ponka, P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol. 2009, 122, 120–133. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)–“2021 update on diagnosis, risk-stratification, and management”. Am. J. Hematol. 2021, 96, 379–394. [Google Scholar] [CrossRef]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Cattivelli, K.; Campagna, D.R.; Schmitz-Abe, K.; Heeney, M.M.; Yaish, H.M.; Caruso Brown, A.E.; Kearney, S.; Walkovich, K.; Markianos, K.; Fleming, M.D.; et al. Ringed sideroblasts in beta-thalassemia. Pediatr. Blood Cancer 2017, 64, e26324. [Google Scholar] [CrossRef]
- Hij, A.; Meunier, A.S.; Vignon, G.; Labrousse, J.; Augereau, P.F.; Carrere, F.; Aucher, P.; Lellouche, F. Rare sideroblastic anemias: About 2 cases, review of the literature and reminder of the main etiologies. Ann. Biol. Clin. 2021, 79, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Falcon, C.P.; Howard, T.H. An infant with Pearson syndrome: A rare cause of congenital sideroblastic anemia and bone marrow failure. Blood 2017, 129, 2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Wiseman, D.H.; May, A.; Jolles, S.; Connor, P.; Powell, C.; Heeney, M.M.; Giardina, P.J.; Klaassen, R.J.; Chakraborty, P.; Geraghty, M.T.; et al. A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood 2013, 122, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.K.; Schmitz-Abe, K.; Kennedy, E.K.; Mamady, H.; Naas, T.; Durie, D.; Campagna, D.R.; Lau, A.; Sendamarai, A.K.; Wiseman, D.H.; et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood 2014, 124, 2867–2871. [Google Scholar] [CrossRef] [Green Version]
- Faraji-Goodarzi, M.; Tarhani, F.; Taee, N. Dyserythropoiesis and myelodysplasia in thiamine-responsive megaloblastic anemia syndrome. Clin. Case Rep. 2020, 8, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Natelson, E.A.; Pyatt, D. Acquired myelodysplasia or myelodysplastic syndrome: Clearing the fog. Adv. Hematol. 2013, 2013, 309637. [Google Scholar] [CrossRef] [Green Version]
- Delmotte, V.; Foidart, P.; De, V.A.; Lejeune, M.; Delwaide, J. Diagnosis of anemia associated with alcoholic cirrhosis. Rev. Med. Liege 2019, 74, 527–534. [Google Scholar]
- Pol, R.R.; Howard, M.R. A man with anemia and a change in personality. Blood 2014, 123, 1784. [Google Scholar] [CrossRef] [Green Version]
- Colucci, G.; Silzle, T.; Solenthaler, M. Pyrazinamide-induced sideroblastic anemia. Am. J. Hematol. 2012, 87, 305. [Google Scholar] [CrossRef] [Green Version]
- Saini, N.; Jacobson, J.O.; Jha, S.; Saini, V.; Weinger, R. The perils of not digging deep enough—Uncovering a rare cause of acquired anemia. Am. J. Hematol. 2012, 87, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willekens, C.; Dumezy, F.; Boyer, T.; Renneville, A.; Rossignol, J.; Berthon, C.; Cotteau-Leroy, A.; Mehiaoui, L.; Quesnel, B.; Preudhomme, C. Linezolid induces ring sideroblasts. Haematologica 2013, 98, e138–e140. [Google Scholar] [CrossRef] [PubMed]
- Liapis, K.; Vrachiolias, G.; Spanoudakis, E.; Kotsianidis, I. Vacuolation of early erythroblasts with ring sideroblasts: A clue to the diagnosis of linezolid toxicity. Br. J. Haematol. 2020, 190, 809. [Google Scholar] [CrossRef] [PubMed]
- Vial, T.; Grignon, M.; Daumont, M.; Guy, C.; Zenut, M.; Germain, M.L.; Jaubert, J.; Ruivard, M.; Guyotat, D.; Descotes, J. Sideroblastic anaemia during fusidic acid treatment. Eur. J. Haematol. 2004, 72, 358–360. [Google Scholar] [CrossRef]
- Ok, C.Y.; Medeiros, L.J.; Hu, Y.; Bueso-Ramos, C.E.; Wang, S.A. Transient/reversible ring sideroblasts in bone marrow of patients post cytotoxic therapies for primary malignancies. Leuk. Res. 2011, 35, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Ramlal, R. “Myelodysplasia” from copper deficiency. Blood 2019, 133, 883. [Google Scholar] [CrossRef]
- Villalba, A.; Senent, L. Differential diagnosis of myelodysplastic syndrome: Anemia associated with copper deficiency. Blood 2018, 131, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. New drugs for myeloid neoplasms with ring sideroblasts: Luspatercept vs imetelstat. Am. J. Hematol. 2021, 96, 761–763. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Platzbecker, U.; Fenaux, P.; Zeidan, A.M.; Garcia-Manero, G.; Mufti, G.J.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Jurcic, J.G.; et al. Luspatercept for myelodysplastic syndromes/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Leukemia 2022, 36, 1432–1435. [Google Scholar] [CrossRef]
- Jouzier, C.; Cherait, A.; Cony-Makhoul, P.; Hamel, J.F.; Veloso, M.; Thepot, S.; Cluzeau, T.; Stamatoullas, A.; Garnier, A.; Guerci-Bresler, A.; et al. Red blood cell transfusion burden in myelodysplastic syndromes (MDS) with ring Sideroblasts (RS): A retrospective multicenter study by the Groupe Francophone des Myelodysplasies (GFM). Transfusion 2022, 62, 961–973. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jadersten, M.; Jansson, M.; Elena, C.; Galli, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Zeinah, G.; DeSancho, M.T. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J. Blood Med. 2020, 11, 305–318. [Google Scholar] [CrossRef] [PubMed]






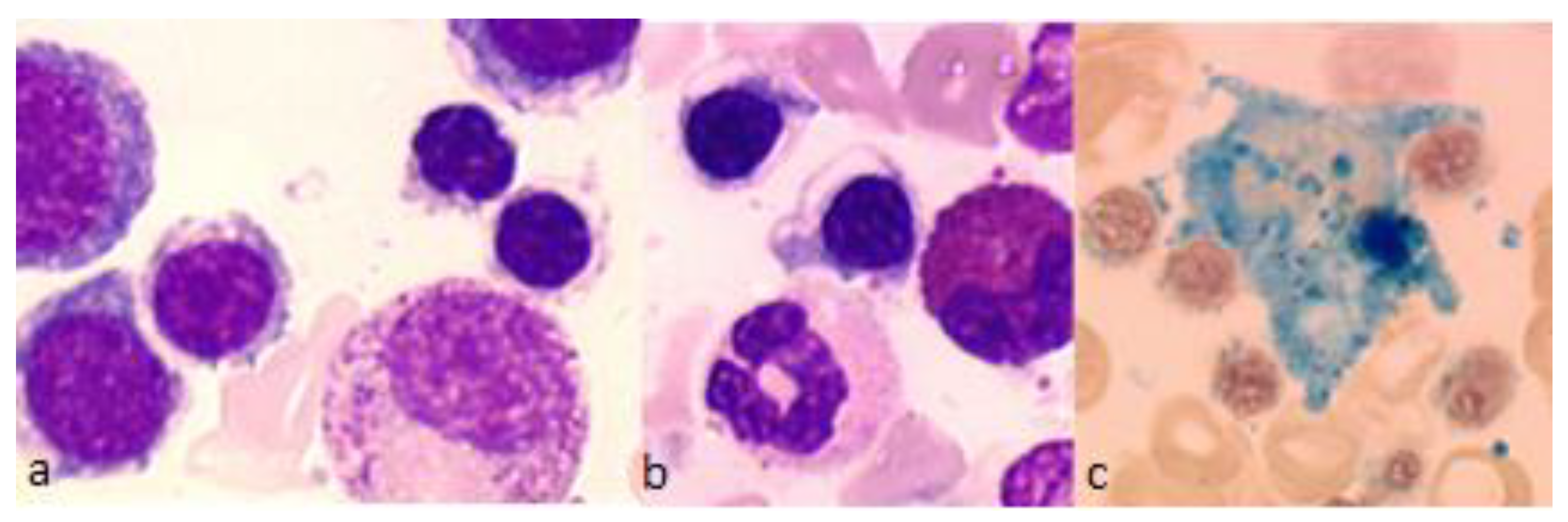{kind=link}
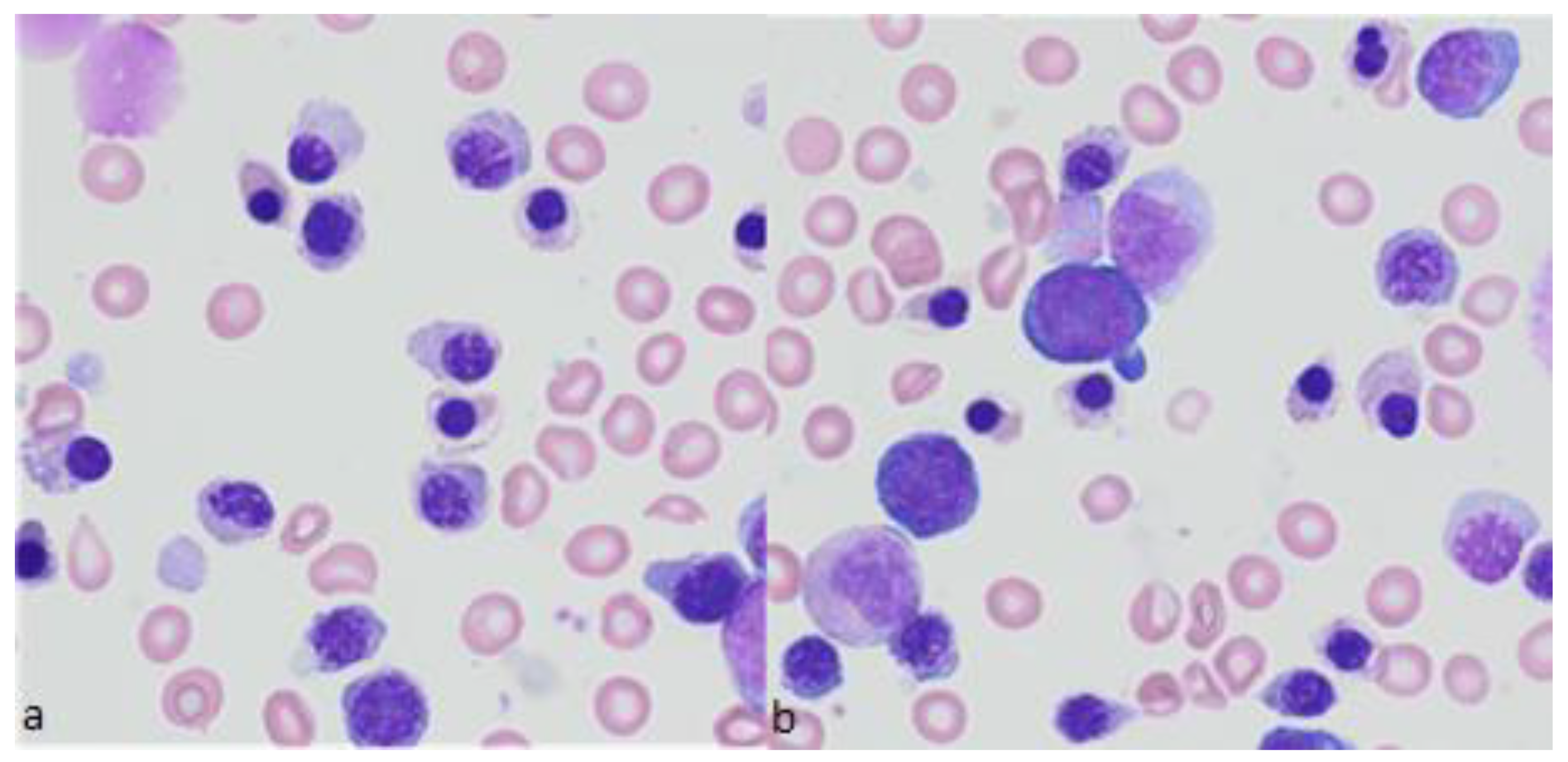{kind=link}
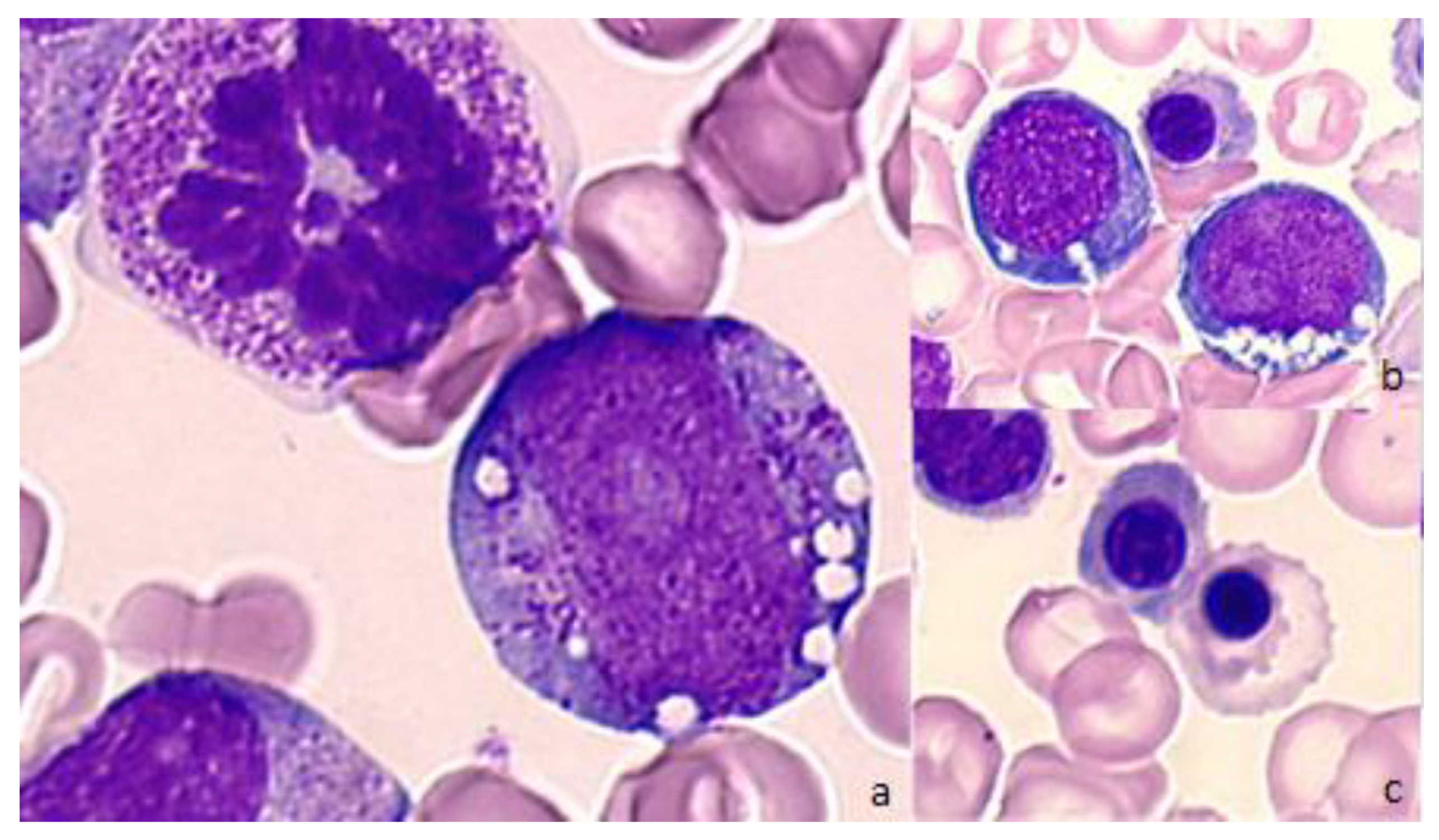{kind=link}
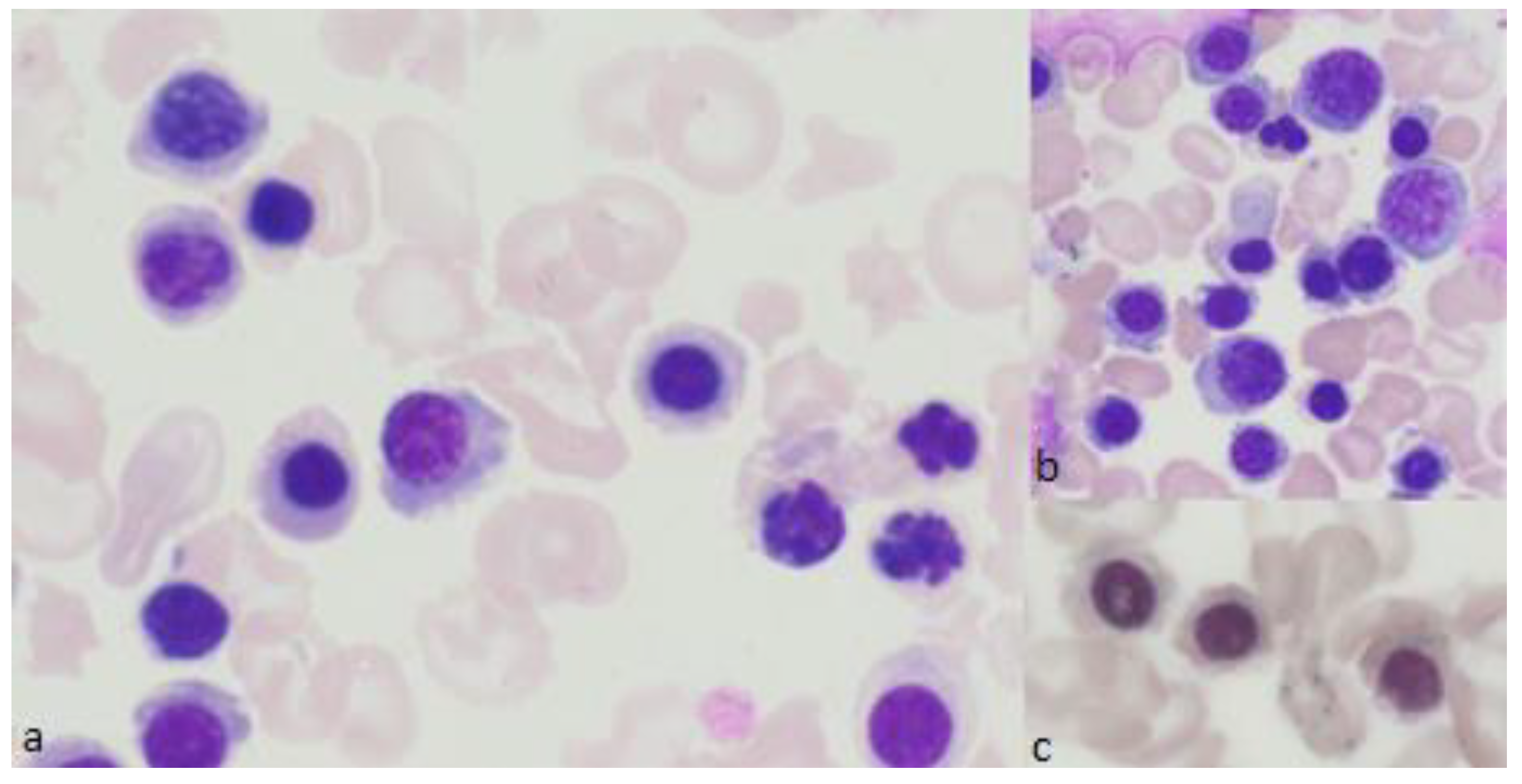{kind=link}
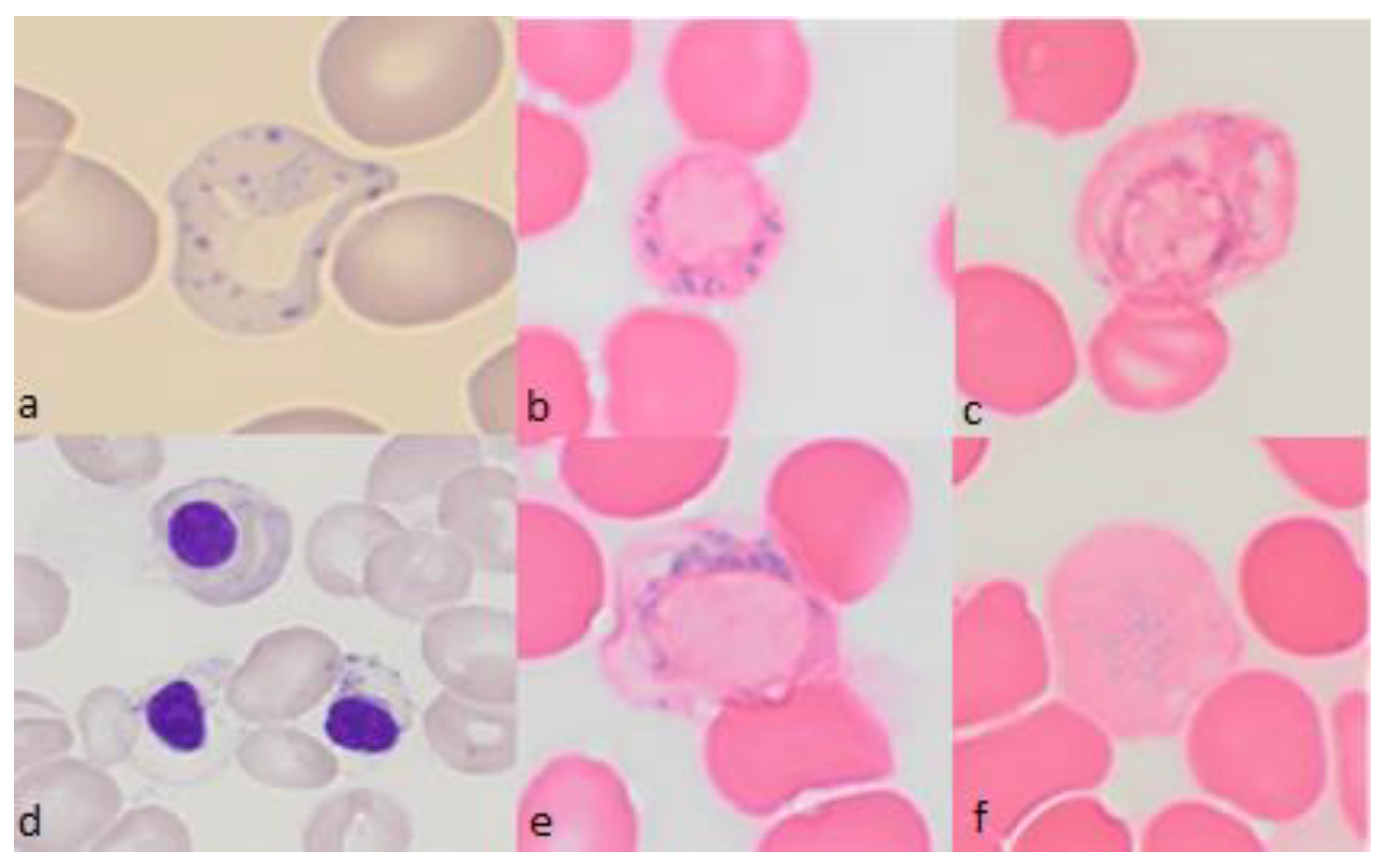{kind=link}
| Condition | Inheritance | Mutations | Biological Features |
|---|---|---|---|
| MDS with low blasts and SF3B1 mutation/MDS with low blasts and ring sideroblasts (MDS-RS: 2016 revised WHO) | Somatic | SF3B1 | Cytopenia of one or two lineages Mild to severe isolated anemia Normal or high mean corpuscular volume Morphological single or multilineage dysplasia <2% blasts in peripheral blood <5% blasts in bone marrow (no Auer rods) Perls’ stain: ≥15% RS with wild-type SF3B1 or ≥5% RS in the presence of SF3B1 mutation |
| MDS/MPN with SF3B1 mutation and thrombocytosis (MDS/MPN -RS-T: 2016 revised WHO) | Somatic | SF3B1 +/− JAK2 V617F | Persistent thrombocytosis (≥450 giga/L) Megakaryocytic hyperplasia and morphological abnormalities MPN like Moderate anemia with erythroid lineage dysplasia, with or without multilineage dysplasia Normal or high mean corpuscular volume <1% blasts in peripheral blood <5% blasts in bone marrow Perls’ stain: ≥15% RS with or without SF3B1 mutation |
| Condition | Gene Anomaly/Chromosomal Localization |
|---|---|
| Non-syndromic sideroblastic anemias | |
| Heme synthesis defects | |
| XLSA = X-linked sideroblastic anemia | ALA synthase gene (ALAS2)—Xp11.21 |
| SIDBA2 = Autosomal recessive pyridoxine refractory sideroblastic anemia | SLC25A38—3p22.1 |
| EPP = Erythropoietic protoporphyria | FECH—18q22 |
| Fe-S biogenesis defects | |
| GLRX5 deficiency | GLRX5—14q32 |
| HSPA9 deficiency | HSPA9—5q31.2 |
| HSCB deficiency | HSCB—22q12.1 |
| Syndromic sideroblastic anemias | |
| Fe-S biogenesis defects | |
| XLSA/A = X-linked sideroblastic anemia and spinocerebellar ataxia | ABCB7—Xq13.1-q13.3 |
| Mitochondrial protein synthesis defects | |
| MLASA1 = Myopathy, lactic acidosis and sideroblastic anemia | YARS2—12p11.21 |
| MLASA2 = Myopathy, lactic acidosis and sideroblastic anemia | PUS1—12q24.33 |
| LARS2 deficiency | LARS2—3p21.3 |
| SFID syndrome | TRNT1—3p26.1 |
| Pearson syndrome | Mitochondrial DNA deletion |
| Mitochondrial respiratory protein mutations | |
| TRMA = Thiamine-responsive megaloblastic anemia | SLC19A2—1q23.3 |
| MT-ATP6-sideroblastic anemia | MT-ATP6 |
| NDUFB11—sideroblastic anemia | NDUFB11–X p11.23 |
| Acquired Reversible Sideroblastic Anemias | |
|---|---|
| Exposure to toxic substances | Alcoholism Heavy metal intoxication (lead, arsenic, mercury) Benzene exposure |
| Drugs | Anti-tuberculosis (isoniazid, pyrazinamide, cycloserine) Antibiotics (chloramphenicol D-penicillamine, linezolid, lincomycin, cefadroxil, fusidic acid, tetracyclines) Cancer chemotherapy (chlorambucil, busulfan, melphalan, lenalidomide) |
| Malnutrition/deficiency in nutrition or other metabolic disorders | Vitamin B1, B6, B9, and B12 deficiencies Copper deficiency Zinc overdose Prolonged parenteral nutrition Gastric surgery Hypothermia |
| Acquired clonal sideroblastic anemias = myelodysplastic neoplasms (or MDS/MPN neoplasms) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girard, S.; Genevieve, F.; Rault, E.; Fenneteau, O.; Lesesve, J.-F. When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics 2022, 12, 1752. https://doi.org/10.3390/diagnostics12071752
Girard S, Genevieve F, Rault E, Fenneteau O, Lesesve J-F. When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics. 2022; 12(7):1752. https://doi.org/10.3390/diagnostics12071752
Chicago/Turabian StyleGirard, Sandrine, Franck Genevieve, Emmanuelle Rault, Odile Fenneteau, and Jean-François Lesesve. 2022. "When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms" Diagnostics 12, no. 7: 1752. https://doi.org/10.3390/diagnostics12071752
APA StyleGirard, S., Genevieve, F., Rault, E., Fenneteau, O., & Lesesve, J. -F. (2022). When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics, 12(7), 1752. https://doi.org/10.3390/diagnostics12071752