
Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection

 ,
,  and
and
Abstract
:1. Introduction
2. Drivers of TAA Formation: A Constant Journey through Gene Discovery
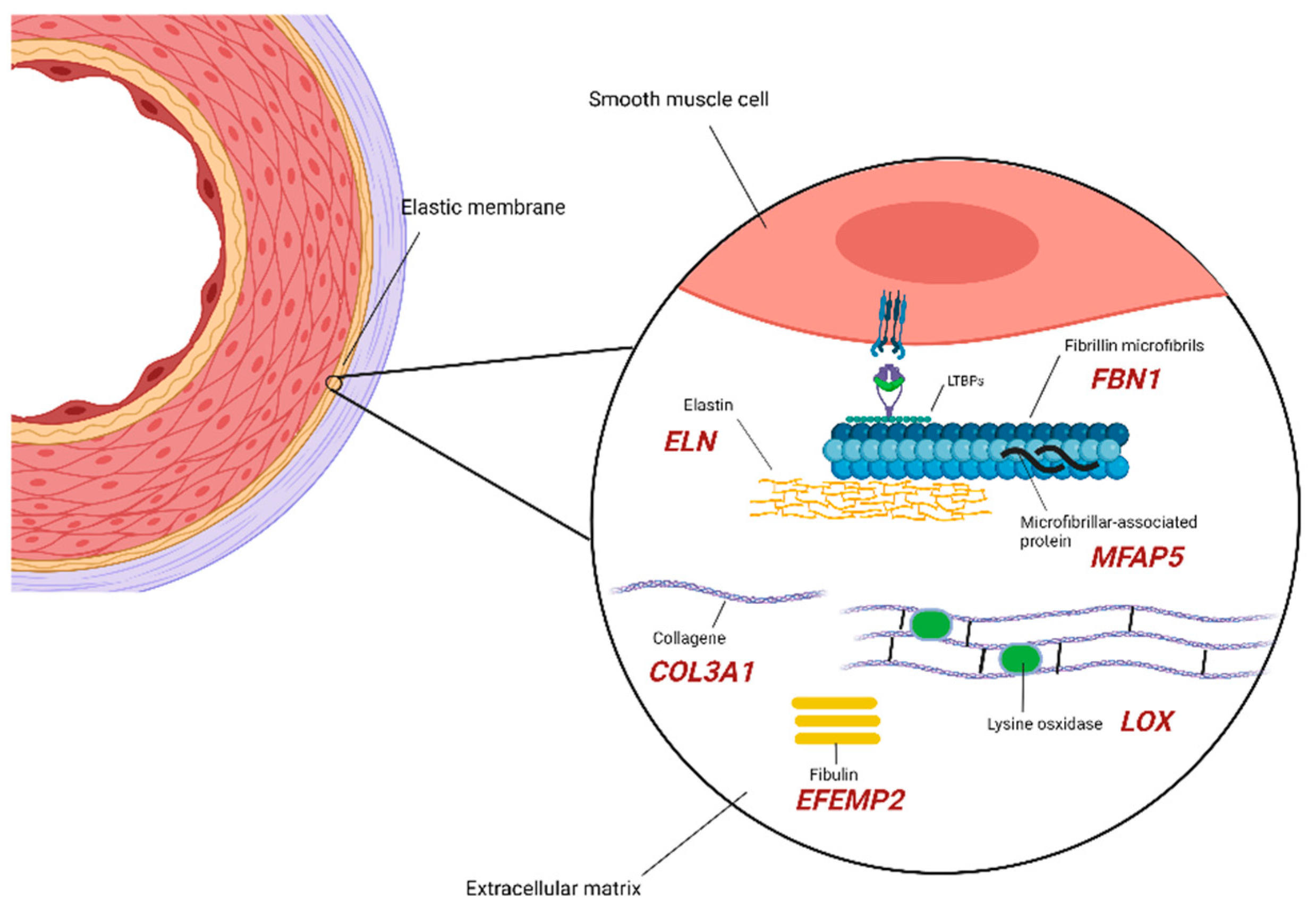
2.1. Extracellular Matrix Components
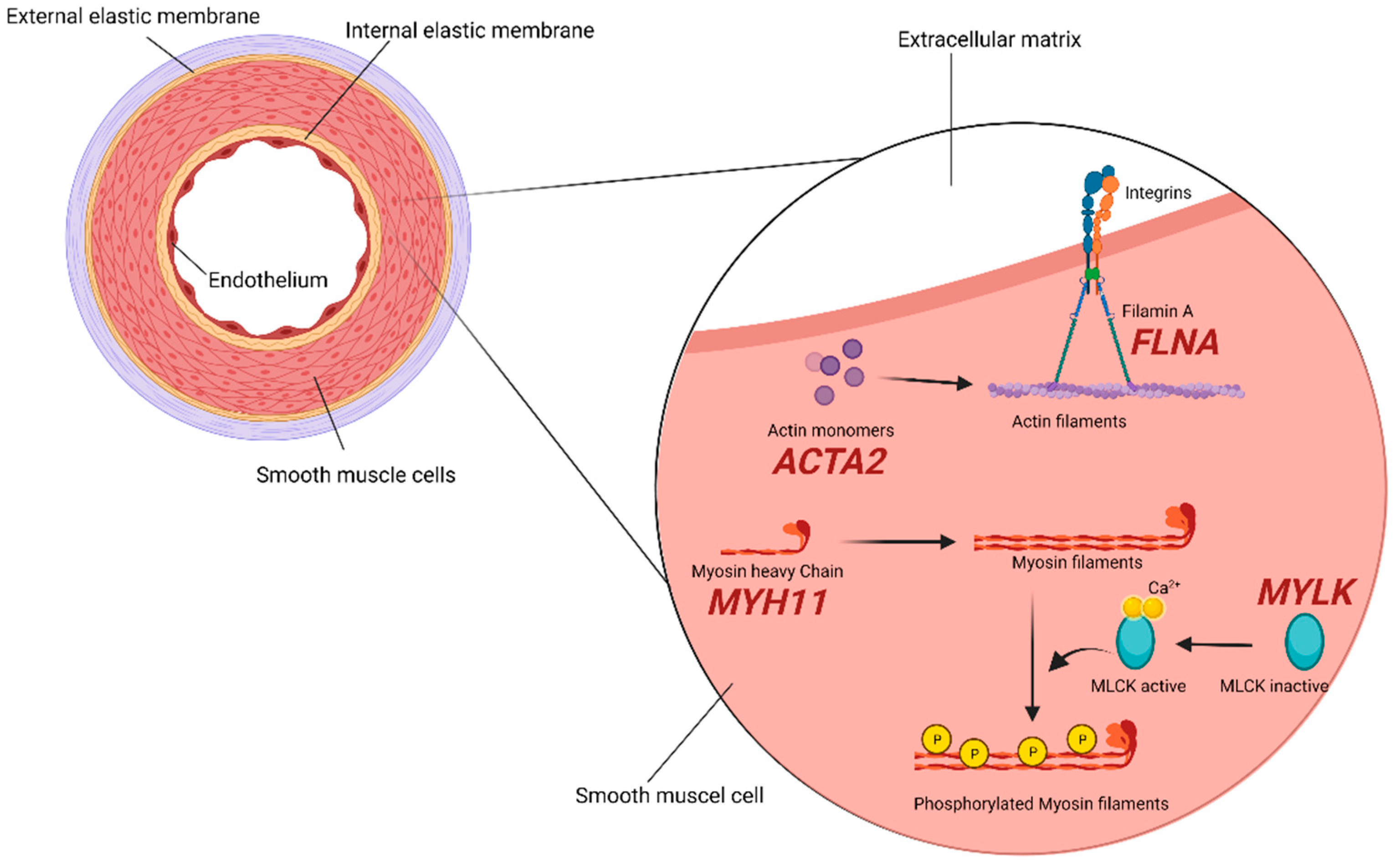
2.2. SMCs (Smooth Muscle Cells) Compartment
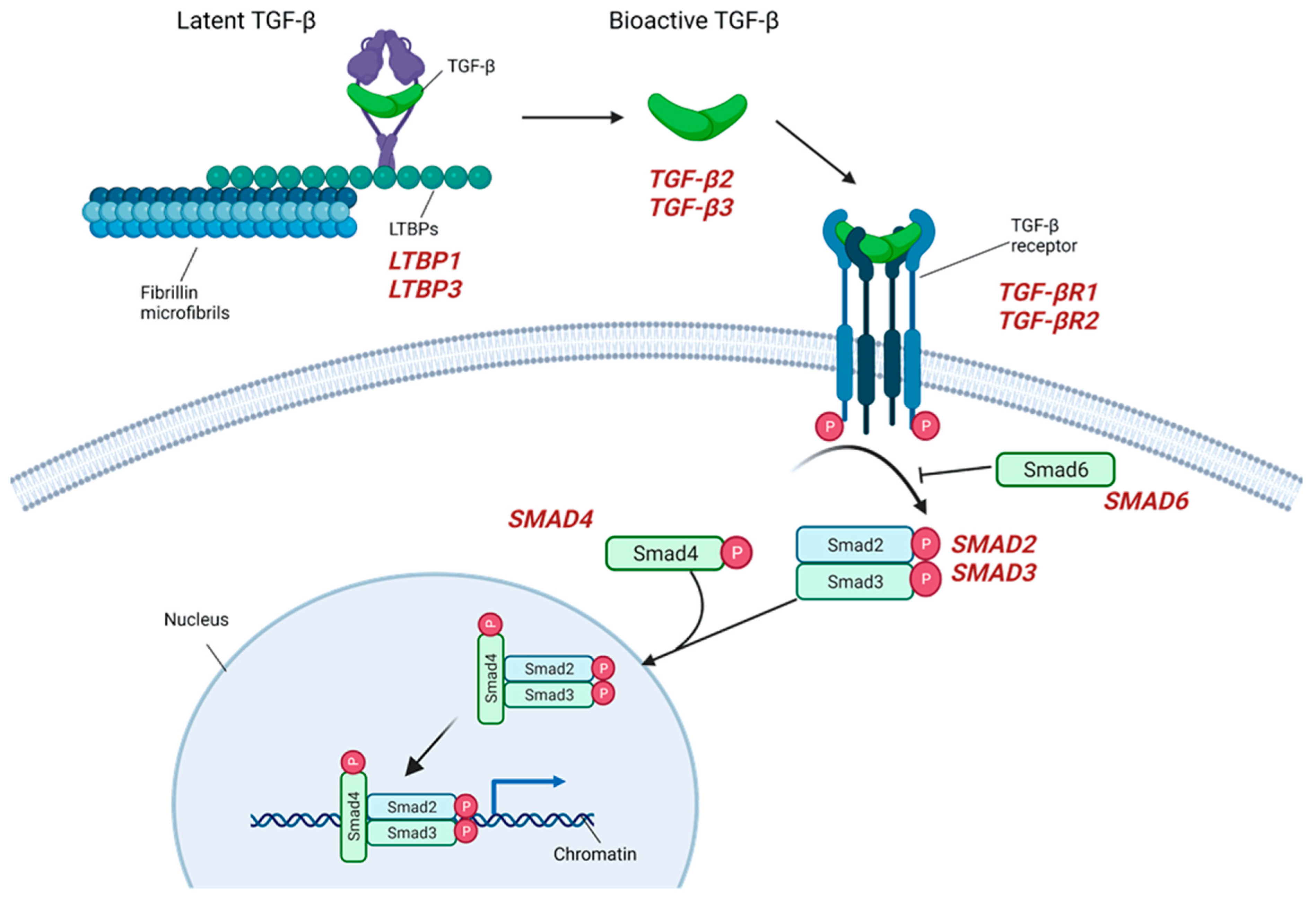
2.3. TGF-β Signaling
2.4. TAA in the Context of Bicuspid Aortic Valve and the Role of Proteases
3. Mechanisms of TAA Progression: The Dissection Menace
3.1. Pathophysiology and Risk Factors
3.2. Genetic Profiles of Dissection
4. Recommended Laboratory Workup for TAA/D Diagnosis and Risk Prediction
4.1. Medical History and Physical Examination
4.2. “Traditional” Circulating Biomarkers for TAA/D
4.3. Genetic Testing in Supporting TAA/D Diagnostics and in Risk Prediction: Where Do We Stand?
5. Latest Findings on TAA/D Genetic and Non-Genetic Biomarkers
5.1. RNA Signatures: A Novel, Noninvasive, and Promising Screening Option?

{kind=link}
{kind=link}
{kind=link}
| Marker | Animal Models | Human Cohort | TAA | TAAD |
|---|---|---|---|---|
| miR-1 | - | aortic tissue specimens from ascending TAA patients (30)/3 tissues of patients with AAA, 11 tissues of patients with TAA and 8 controls [185,186] | + | |
| miR-21 | + | |||
| miR-29a | + | + | ||
| miR-133a | + | + | ||
| miR-15a | - | 10 patients with TAA/3 tissue specimens from AAA patients, 11 from TAA patients and 8 controls/aortic tissue specimens from AAA patients (10) [186,187,188] | + | + |
| miR-22 | + | + | ||
| miR-25 | + | |||
| miR-29b | + | |||
| miR-125a-3p | + | |||
| miR-126-3p | + | |||
| miR-128 | + | |||
| miR-133b | + | + | ||
| miR-138-1 | + | + | ||
| miR-142–5p | + | |||
| miR-145 | + | + | ||
| miR-146b-5p | + | |||
| miR-183 | + | + | ||
| miR-422a | + | |||
| miR-433 | + | + | ||
| miR-486–5p | + | |||
| miR-487b | + | |||
| miR-491–3p | + | + | ||
| miR-553 | + | + | ||
| miR-638 | + | |||
| miR-940 | + | + | ||
| miR-193a-3p | + | + | ||
| miR-768–5p | + | + | ||
| miR-886–5p | + | + | ||
| miR-195 | + | + | ||
| miR-140–5p | + | + | ||
| miR-30e | + | + | ||
| miR-101 | + | + | ||
| miR-744 | + | + | ||
| miR-193a-5p | + | + | ||
| miR-30c | - | 3 tissues specimens from AAA patients, 11 from TAA patients and 8 controls [186] | + | |
| miR-155 | + | |||
| miR-204 | + | |||
| miR-143 | mouse models [183] | - | + | + |
5.2. Novel Genes and the WES Outbreak: Pros and Cons in the Clinical Practice and Applicability
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| AAAD | acute Stanford type A aortic dissection |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin motifs |
| BAV | bicuspid aortic valve |
| circRNAs | circular RNAs |
| CK-MM | Creatine-kinase isozyme MM |
| CRP | C-reactive protein |
| DD | d-Dimer |
| ECM | extracellular matrix |
| ESC | European Society of Cardiology |
| FTAAD | familial thoracic aortic aneurysm and dissection |
| Hcy | homocysteine |
| LAP | latency-associated peptide |
| LDS | Loeys-Dietz syndrome |
| lncRNAs | long non-coding RNAs |
| LTBP | latent TGF? binding protein |
| MFS | Marfan syndrome |
| miRNAs | micro-RNAs |
| MMPs | metalloproteinases |
| MPV | mean platelet volume |
| ncRNAs | non-coding RNAs |
| NGS | next-generation sequencing |
| PC1 | Polycistin 1 |
| piRNAs | Piwi-interacting RNAs |
| PLT | platelet |
| qRT-PCR | Quantitative Reverse Transcription Polymerase Chain Reaction |
| rasiRNAs | repeat associated small interfering RNAs |
| sELAFs | soluble elastin fragments |
| SMCs | smooth muscle cells |
| smMHC | smooth muscle myosin heavy chain |
| TAA/D | thoracic aortic aneurysm and dissection |
| TAA | thoracic aortic aneurys |
| TAD | thoracic aortic dissection |
| TEVAR | thoracic endovascular aortic repair |
| TGFβ | Transforming Growth Factor-β |
| tHcy | total homocysteine |
| TIMPs | tissue inhibitors of metalloproteinases |
| vEDS | vascular Ehlers-Danlos syndrome |
| VEGF | vascular endothelial growth factor |
| VSMCs | vascular smooth muscle cells |
| VUS | variant of uncertain significance |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Bossone, E.; Eagle, K.A. Epidemiology and Management of Aortic Disease: Aortic Aneurysms and Acute Aortic Syndromes. Nat. Rev. Cardiol. 2021, 18, 331–348. [Google Scholar] [CrossRef]
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural History of Thoracic Aortic Aneurysms. J. Vasc. Surg. 2012, 56, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouse, W.D.; Hallett, J.W.; Schaff, H.V.; Spittell, P.C.; Rowland, C.M.; Ilstrup, D.M.; Melton, L.J. Acute Aortic Dissection: Population-Based Incidence Compared with Degenerative Aortic Aneurysm Rupture. Mayo Clin. Proc. 2004, 79, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Nienaber, C.A.; Clough, R.E.; Sakalihasan, N.; Suzuki, T.; Gibbs, R.; Mussa, F.; Jenkins, M.P.; Thompson, M.M.; Evangelista, A.; Yeh, J.S.M.; et al. Aortic Dissection. Nat. Rev. Dis. Primer 2016, 2, 16053. [Google Scholar] [CrossRef] [PubMed]
- Smedberg, C.; Steuer, J.; Leander, K.; Hultgren, R. Sex Differences and Temporal Trends in Aortic Dissection: A Population-Based Study of Incidence, Treatment Strategies, and Outcome in Swedish Patients during 15 Years. Eur. Heart J. 2020, 41, 2430–2438. [Google Scholar] [CrossRef] [PubMed]
- Faggion Vinholo, T.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Nonsyndromic Thoracic Aortic Aneurysms and Dissections—Is Screening Possible? Semin. Thorac. Cardiovasc. Surg. 2019, 31, 628–634. [Google Scholar] [CrossRef]
- Salameh, M.J.; Black, J.H.; Ratchford, E.V. Thoracic Aortic Aneurysm. Vasc. Med. 2018, 23, 573–578. [Google Scholar] [CrossRef]
- Monda, E.; Fusco, A.; Della Corte, A.; Caiazza, M.; Cirillo, A.; Gragnano, F.; Giugliano, M.P.; Citro, R.; Rubino, M.; Esposito, A.; et al. Impact of Regular Physical Activity on Aortic Diameter Progression in Paediatric Patients with Bicuspid Aortic Valve. Pediatr. Cardiol. 2021, 42, 1133–1140. [Google Scholar] [CrossRef]
- Rohde, S.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Thoracic Aortic Aneurysm Gene Dictionary. Asian Cardiovasc. Thorac. Ann. 2021, 29, 682–696. [Google Scholar] [CrossRef]
- Chou, E.L.; Lindsay, M.E. The Genetics of Aortopathies: Hereditary Thoracic Aortic Aneurysms and Dissections. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 136–148. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Di Bartolomeo, R.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases. Kardiol. Pol. 2014, 72, 1169–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Lindsay, M.E. Role of Clinical Genetic Testing in the Management of Aortopathies. Curr. Cardiol. Rep. 2021, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Elefteriades, J.A.; Sang, A.; Kuzmik, G.; Hornick, M. Guilt by Association: Paradigm for Detecting a Silent Killer (Thoracic Aortic Aneurysm). Open Heart 2015, 2, e000169. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial Thoracic Aortic Aneurysms and Dissections—Incidence, Modes of Inheritance, and Phenotypic Patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial Thoracic Aortic Dilatations and Dissections: A Case Control Study. J. Vasc. Surg. 1997, 25, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial Patterns of Thoracic Aortic Aneurysms. Arch. Surg. 1999, 134, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Cannon Albright, L.A.; Camp, N.J.; Farnham, J.M.; MacDonald, J.; Abtin, K.; Rowe, K.G. A Genealogical Assessment of Heritable Predisposition to Aneurysms. J. Neurosurg. 2003, 99, 637–643. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-Function Mutations in the X-Linked Biglycan Gene Cause a Severe Syndromic Form of Thoracic Aortic Aneurysms and Dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; Komuro, I. Genetic Basis of Hereditary Thoracic Aortic Aneurysms and Dissections. J. Cardiol. 2019, 74, 136–143. [Google Scholar] [CrossRef]
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic Fibres in Health and Disease. Expert Rev. Mol. Med. 2013, 15, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, Z. Aortic Aneurysmal Disease and Cutis Laxa Caused by Defects in the Elastin Gene. J. Med. Genet. 2005, 43, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guemann, A.-S.; Andrieux, J.; Petit, F.; Halimi, E.; Bouquillon, S.; Manouvrier-Hanu, S.; Van De Kamp, J.; Boileau, C.; Hanna, N.; Jondeau, G.; et al. ELN Gene Triplication Responsible for Familial Supravalvular Aortic Aneurysm. Cardiol. Young 2015, 25, 712–717. [Google Scholar] [CrossRef]
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.M.; Dietz, H.C. Genotype-Phenotype Correlation in Patients with Bicuspid Aortic Valve and Aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146, 158–165.e1. [Google Scholar] [CrossRef] [Green Version]
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted Genetic Analysis in a Large Cohort of Familial and Sporadic Cases of Aneurysm or Dissection of the Thoracic Aorta. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.A.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J.M. Genotype Impacts Survival in Marfan Syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef]
- Guo, D.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.P.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ. Res. 2016, 118, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Brigham Genomic Medicine; Mecham, R.P.; et al. Loss of Function Mutation in LOX Causes Thoracic Aortic Aneurysm and Dissection in Humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.-Q.; Medina-Martinez, O.; Guo, D.-C.; Gong, L.; Regalado, E.S.; Reynolds, C.L.; Boileau, C.; Jondeau, G.; Prakash, S.K.; Kwartler, C.S.; et al. FOXE3 Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. J. Clin. Investig. 2016, 126, 948–961. [Google Scholar] [CrossRef] [Green Version]
- Schubert, J.A.; Landis, B.J.; Shikany, A.R.; Hinton, R.B.; Ware, S.M. Clinically Relevant Variants Identified in Thoracic Aortic Aneurysm Patients by Research Exome Sequencing. Am. J. Med. Genet. A 2016, 170, 1288–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.; Chang, B.; Walsh, C.A. Thoracic Aortic Aneurysm in Patients with Loss of Function Filamin A Mutations: Clinical Characterization, Genetics, and Recommendations. Am. J. Med. Genet. A. 2018, 176, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in Myosin Heavy Chain 11 Cause a Syndrome Associating Thoracic Aortic Aneurysm/Aortic Dissection and Patent Ductus Arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Luyckx, I.; Loeys, B.L. Curriculum topic: Disease of the aorta and trauma to the aorta and heart The Genetic Architecture of Non-Syndromic Thoracic Aortic Aneurysm. Heart Br. Card. Soc. 2015, 101, 1678–1684. [Google Scholar] [CrossRef]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- Ponińska, J.K.; Bilińska, Z.T.; Truszkowska, G.; Michalak, E.; Podgórska, A.; Stępień-Wojno, M.; Chmielewski, P.; Lutyńska, A.; Płoski, R. Good Performance of the Criteria of American College of Medical Genetics and Genomics/Association for Molecular Pathology in Prediction of Pathogenicity of Genetic Variants Causing Thoracic Aortic Aneurysms and Dissections. J. Transl. Med. 2022, 20, 42. [Google Scholar] [CrossRef]
- Quiñones-Pérez, B.; VanNoy, G.E.; Towne, M.C.; Shen, Y.; Singh, M.N.; Agrawal, P.B.; Smith, S.E. Three-Generation Family with Novel Contiguous Gene Deletion on Chromosome 2p22 Associated with Thoracic Aortic Aneurysm Syndrome. Am. J. Med. Genet. A 2018, 176, 560–569. [Google Scholar] [CrossRef]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys–Dietz Syndrome: A Primer for Diagnosis and Management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Regalado, E. Heritable Thoracic Aortic Disease Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Wooten, E.C.; Iyer, L.K.; Montefusco, M.C.; Hedgepeth, A.K.; Payne, D.D.; Kapur, N.K.; Housman, D.E.; Mendelsohn, M.E.; Huggins, G.S. Application of Gene Network Analysis Techniques Identifies AXIN1/PDIA2 and Endoglin Haplotypes Associated with Bicuspid Aortic Valve. PLoS ONE 2010, 5, e8830. [Google Scholar] [CrossRef]
- Takeda, N.; Morita, H.; Fujita, D.; Inuzuka, R.; Taniguchi, Y.; Imai, Y.; Hirata, Y.; Komuro, I. Congenital Contractural Arachnodactyly Complicated with Aortic Dilatation and Dissection: Case Report and Review of Literature. Am. J. Med. Genet. A 2015, 167, 2382–2387. [Google Scholar] [CrossRef]
- Guo, D.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A Mutations Predispose Individuals to Thoracic Aortic Aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.; De Paepe, A.; Coucke, P. Arterial Tortuosity Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lillie, M.A.; David, G.J.; Gosline, J.M. Mechanical Role of Elastin-Associated Microfibrils in Pig Aortic Elastic Tissue. Connect. Tissue Res. 1998, 37, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes 2021, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, C.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan Syndrome Caused by a Recurrent de Novo Missense Mutation in the Fibrillin Gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Csiszar, K. Lysyl Oxidases: A Novel Multifunctional Amine Oxidase Family. In Progress in Nucleic Acid Research and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 70l, pp. 1–32. ISBN 978-0-12-540070-1. [Google Scholar]
- Zentner, D.; James, P.; Bannon, P.; Jeremy, R. Familial Aortopathies—State of the Art Review. Heart Lung Circ. 2020, 29, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Papke, C.L.; Yanagisawa, H. Fibulin-4 and Fibulin-5 in Elastogenesis and beyond: Insights from Mouse and Human Studies. Matrix Biol. 2014, 37, 142–149. [Google Scholar] [CrossRef]
- De Figueiredo Borges, L.; Jaldin, R.G.; Dias, R.R.; Stolf, N.A.G.; Michel, J.-B.; Gutierrez, P.S. Collagen Is Reduced and Disrupted in Human Aneurysms and Dissections of Ascending Aorta. Hum. Pathol. 2008, 39, 437–443. [Google Scholar] [CrossRef]
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-Associated Glycoprotein 2 (MAGP2) Loss of Function Has Pleiotropic Effects in Vivo. J. Biol. Chem. 2013, 288, 28869–28880. [Google Scholar] [CrossRef] [Green Version]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans Decorin and Biglycan Differentially Modulate TGF-β-Mediated Fibrotic Responses in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [CrossRef]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in Smooth Muscle α-Actin (ACTA2) Lead to Thoracic Aortic Aneurysms and Dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Mutation of ACTA2 Gene as an Important Cause of Familial and Nonfamilial Nonsyndromatic Thoracic Aortic Aneurysm and/or Dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of Dissection in Thoracic Aneurysms Associated with Mutations of Smooth Muscle Alpha-Actin 2 (ACTA2). Heart Br. Card. Soc. 2011, 97, 321–326. [Google Scholar] [CrossRef]
- Lu, H.; Fagnant, P.M.; Bookwalter, C.S.; Joel, P.; Trybus, K.M. Vascular Disease-Causing Mutation R258C in ACTA2 Disrupts Actin Dynamics and Interaction with Myosin. Proc. Natl. Acad. Sci. USA 2015, 112, E4168–E4177. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; McCulloch, C.A. Filamin A Mediates Interactions between Cytoskeletal Proteins That Control Cell Adhesion. FEBS Lett. 2011, 585, 18–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.T.; Fisher, S.D. Heritable FLNA Gene Mutation in a Patient with Thoracic Aortic Aneurysm. JACC Case Rep. 2022, 4, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Pomianowski, P.; Elefteriades, J.A. The Genetics and Genomics of Thoracic Aortic Disease. Ann. Cardiothorac. Surg. 2013, 2, 271–279. [Google Scholar] [CrossRef]
- Daugherty, A.; Chen, Z.; Sawada, H.; Rateri, D.L.; Sheppard, M.B. Transforming Growth Factor-β in Thoracic Aortic Aneurysms: Good, Bad, or Irrelevant? J. Am. Heart Assoc. 2017, 6, e005221. [Google Scholar] [CrossRef]
- Michel, J.-B.; Jondeau, G.; Milewicz, D.M. From Genetics to Response to Injury: Vascular Smooth Muscle Cells in Aneurysms and Dissections of the Ascending Aorta. Cardiovasc. Res. 2018, 114, 578–589. [Google Scholar] [CrossRef]
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; Weissler, K.; et al. Lineage-Specific Events Underlie Aortic Root Aneurysm Pathogenesis in Loeys-Dietz Syndrome. J. Clin. Investig. 2019, 129, 659–675. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Dietz, H.C. Lessons on the Pathogenesis of Aneurysm from Heritable Conditions. Nature 2011, 473, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.-C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A Mutation Update on the LDS-Associated Genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecconi, M.; Manfrin, M.; Moraca, A.; Zanoli, R.; Colonna, P.L.; Bettuzzi, M.G.; Moretti, S.; Gabrielli, D.; Perna, G.P. Aortic Dimensions in Patients with Bicuspid Aortic Valve without Significant Valve Dysfunction. Am. J. Cardiol. 2005, 95, 292–294. [Google Scholar] [CrossRef]
- Pepe, G.; Nistri, S.; Giusti, B.; Sticchi, E.; Attanasio, M.; Porciani, C.; Abbate, R.; Bonow, R.O.; Yacoub, M.; Gensini, G.F. Identification of Fibrillin 1 Gene Mutations in Patients with Bicuspid Aortic Valve (BAV) without Marfan Syndrome. BMC Med. Genet. 2014, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Boyum, J.; Fellinger, E.K.; Schmoker, J.D.; Trombley, L.; McPartland, K.; Ittleman, F.P.; Howard, A.B. Matrix Metalloproteinase Activity in Thoracic Aortic Aneurysms Associated with Bicuspid and Tricuspid Aortic Valves. J. Thorac. Cardiovasc. Surg. 2004, 127, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Rabkin, S.W. Differential Expression of MMP-2, MMP-9 and TIMP Proteins in Thoracic Aortic Aneurysm - Comparison with and without Bicuspid Aortic Valve: A Meta-Analysis. Vasa 2014, 43, 433–442. [Google Scholar] [CrossRef]
- Martin-Blazquez, A.; Heredero, A.; Aldamiz-Echevarria, G.; Martin-Lorenzo, M.; Alvarez-Llamas, G. Non-Syndromic Thoracic Aortic Aneurysm: Cellular and Molecular Insights. J. Pathol. 2021, 254, 229–238. [Google Scholar] [CrossRef]
- Geng, L.; Wang, W.; Chen, Y.; Cao, J.; Lu, L.; Chen, Q.; He, R.; Shen, W. Elevation of ADAM10, ADAM17, MMP-2 and MMP-9 Expression with Media Degeneration Features CaCl2-Induced Thoracic Aortic Aneurysm in a Rat Model. Exp. Mol. Pathol. 2010, 89, 72–81. [Google Scholar] [CrossRef]
- Ren, P.; Zhang, L.; Xu, G.; Palmero, L.C.; Albini, P.T.; Coselli, J.S.; Shen, Y.H.; LeMaire, S.A. ADAMTS-1 and ADAMTS-4 Levels Are Elevated in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2013, 95, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Wilton, E.; Bland, M.; Thompson, M.; Jahangiri, M. Matrix Metalloproteinase Expression in the Ascending Aorta and Aortic Valve. Interact. Cardiovasc. Thorac. Surg. 2008, 7, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.; Isselbacher, E.M.; Bossone, E.; Gleason, T.G.; Eusanio, M.D.; Sechtem, U.; Ehrlich, M.P.; Trimarchi, S.; Braverman, A.C.; Myrmel, T.; et al. Insights from the International Registry of Acute Aortic Dissection: A 20-Year Experience of Collaborative Clinical Research. Circulation 2018, 137, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Silaschi, M.; Byrne, J.; Wendler, O. Aortic Dissection: Medical, Interventional and Surgical Management. Heart 2017, 103, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Hagan, P.G.; Nienaber, C.A.; Isselbacher, E.M.; Bruckman, D.; Karavite, D.J.; Russman, P.L.; Evangelista, A.; Fattori, R.; Suzuki, T.; Oh, J.K.; et al. The International Registry of Acute Aortic Dissection (IRAD): New Insights into an Old Disease. JAMA 2000, 283, 897. [Google Scholar] [CrossRef]
- Vilacosta, I.; San Román, J.A.; di Bartolomeo, R.; Eagle, K.; Estrera, A.L.; Ferrera, C.; Kaji, S.; Nienaber, C.A.; Riambau, V.; Schäfers, H.-J.; et al. Acute Aortic Syndrome Revisited: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 2106–2125. [Google Scholar] [CrossRef]
- White, A.; Broder, J.; Mando-Vandrick, J.; Wendell, J.; Crowe, J. Acute Aortic Emergencies—Part 2 Aortic Dissections. Adv. Emerg. Nurs. J. 2013, 35, 28–52. [Google Scholar] [CrossRef]
- Von Kodolitsch, Y.; Csösz, S.K.; Koschyk, D.H.; Schalwat, I.; Loose, R.; Karck, M.; Dieckmann, C.; Fattori, R.; Haverich, A.; Berger, J.; et al. Intramural Hematoma of the Aorta: Predictors of Progression to Dissection and Rupture. Circulation 2003, 107, 1158–1163. [Google Scholar] [CrossRef]
- Manabe, T.; Imoto, K.; Uchida, K.; Doi, C.; Takanashi, Y. Decreased Tissue Inhibitor of Metalloproteinase-2/Matrix Metalloproteinase Ratio in the Acute Phase of Aortic Dissection. Surg. Today 2004, 34, 220–225. [Google Scholar] [CrossRef]
- Koullias, G.J.; Ravichandran, P.; Korkolis, D.P.; Rimm, D.L.; Elefteriades, J.A. Increased Tissue Microarray Matrix Metalloproteinase Expression Favors Proteolysis in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2004, 78, 2106–2110. [Google Scholar] [CrossRef]
- Del Porto, F.; di Gioia, C.; Tritapepe, L.; Ferri, L.; Leopizzi, M.; Nofroni, I.; De Santis, V.; Della Rocca, C.; Mitterhofer, A.P.; Bruno, G.; et al. The Multitasking Role of Macrophages in Stanford Type A Acute Aortic Dissection. Cardiology 2014, 127, 123–129. [Google Scholar] [CrossRef]
- Landenhed, M.; Engström, G.; Gottsäter, A.; Caulfield, M.P.; Hedblad, B.; Newton-Cheh, C.; Melander, O.; Smith, J.G. Risk Profiles for Aortic Dissection and Ruptured or Surgically Treated Aneurysms: A Prospective Cohort Study. J. Am. Heart Assoc. 2015, 4, e001513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.W.A.; Jonas, U.; Bühler, F.R.; Resink, T.J. Activation of Human Peripheral Monocytes by Angiotensin II. FEBS Lett. 1994, 347, 178–180. [Google Scholar] [CrossRef] [Green Version]
- Stumpf, C.; Jukic, J.; Yilmaz, A.; Raaz, D.; Schmieder, R.E.; Daniel, W.G.; Garlichs, C.D. Elevated VEGF-Plasma Levels in Young Patients with Mild Essential Hypertension. Eur. J. Clin. Investig. 2009, 39, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; D’Angelo, A.; Ciccarelli, L.; Piccinni, M.N.; Pricolo, F.; Salvadeo, S.; Montagna, L.; Gravina, A.; Ferrari, I.; Galli, S.; et al. Matrix Metalloproteinase-2, -9, and Tissue Inhibitor of Metalloproteinase-1 in Patients with Hypertension. Endothel. J. Endothel. Cell Res. 2006, 13, 227–231. [Google Scholar] [CrossRef]
- Elefteriades, J.A. Natural History of Thoracic Aortic Aneurysms: Indications for Surgery, and Surgical versus Nonsurgical Risks. Ann. Thorac. Surg. 2002, 74, S1877–S1880, discussion S1892–S1898. [Google Scholar] [CrossRef]
- Pape, L.A.; Tsai, T.T.; Isselbacher, E.M.; Oh, J.K.; O’gara, P.T.; Evangelista, A.; Fattori, R.; Meinhardt, G.; Trimarchi, S.; Bossone, E.; et al. Aortic Diameter > or =5.5 Cm Is Not a Good Predictor of Type A Aortic Dissection: Observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007, 116, 1120–1127. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.R.; Goldstein, L.J.; Coady, M.A.; Tittle, S.L.; Rizzo, J.A.; Kopf, G.S.; Elefteriades, J.A. Yearly Rupture or Dissection Rates for Thoracic Aortic Aneurysms: Simple Prediction Based on Size. Ann. Thorac. Surg. 2002, 73, 17–27, discussion 27–28. [Google Scholar] [CrossRef]
- Gawinecka, J.; Schönrath, F.; von Eckardstein, A. Acute Aortic Dissection: Pathogenesis, Risk Factors and Diagnosis. Swiss Med. Wkly. 2017, 147, w14489. [Google Scholar] [CrossRef]
- Pyeritz, R.E. Recent Progress in Understanding the Natural and Clinical Histories of the Marfan Syndrome. Trends Cardiovasc. Med. 2016, 26, 423–428. [Google Scholar] [CrossRef]
- Weinsaft, J.W.; Devereux, R.B.; Preiss, L.R.; Feher, A.; Roman, M.J.; Basson, C.T.; Geevarghese, A.; Ravekes, W.; Dietz, H.C.; Holmes, K.; et al. Aortic Dissection in Patients with Genetically Mediated Aneurysms. J. Am. Coll. Cardiol. 2016, 67, 2744–2754. [Google Scholar] [CrossRef]
- Den Hartog, A.W.; Franken, R.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; de Waard, V.; Pals, G.; Mulder, B.J.M.; Groenink, M. The Risk for Type B Aortic Dissection in Marfan Syndrome. J. Am. Coll. Cardiol. 2015, 65, 246–254. [Google Scholar] [CrossRef]
- Pape, L.A.; Awais, M.; Woznicki, E.M.; Suzuki, T.; Trimarchi, S.; Evangelista, A.; Myrmel, T.; Larsen, M.; Harris, K.M.; Greason, K.; et al. Presentation, Diagnosis, and Outcomes of Acute Aortic Dissection: 17-Year Trends from the International Registry of Acute Aortic Dissection. J. Am. Coll. Cardiol. 2015, 66, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Tran-Fadulu, V.; Pannu, H.; Kim, D.H.; Vick, G.W.; Lonsford, C.M.; Lafont, A.L.; Boccalandro, C.; Smart, S.; Peterson, K.L.; Hain, J.Z.; et al. Analysis of Multigenerational Families with Thoracic Aortic Aneurysms and Dissections Due to TGFBR1 or TGFBR2 Mutations. J. Med. Genet. 2009, 46, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muiño-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ. Cardiovasc. Genet. 2016, 9, 548–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.-A.; Venselaar, H.; et al. Mutations in SMAD3 Cause a Syndromic Form of Aortic Aneurysms and Dissections with Early-Onset Osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Laterza, D.; Ritelli, M.; Zini, A.; Colombi, M.; Dell’Acqua, M.L.; Vandelli, L.; Bigliardi, G.; Verganti, L.; Vallone, S.; Vincenzi, C.; et al. Novel Pathogenic TGFBR1 and SMAD3 Variants Identified after Cerebrovascular Events in Adult Patients with Loeys-Dietz Syndrome. Eur. J. Med. Genet. 2019, 62, 103727. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Pan, X.; Li, W.; Sun, L.; Guo, J. Identification of Clinically Relevant Variants by Whole Exome Sequencing in Chinese Patients with Sporadic Non-Syndromic Type A Aortic Dissection. Clin. Chim. Acta 2020, 506, 160–165. [Google Scholar] [CrossRef]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.-C.; Hanna, N.; Arnaud, P.; Muiño-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 Pathogenic Variants: Risk for Thoracic Aortic Disease and Associated Complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260. [Google Scholar] [CrossRef]
- Hilhorst-Hofstee, Y.; Scholte, A.J.H.A.; Rijlaarsdam, M.E.B.; van Haeringen, A.; Kroft, L.J.; Reijnierse, M.; Ruivenkamp, C.A.L.; Versteegh, M.I.M.; Pals, G.; Breuning, M.H. An Unanticipated Copy Number Variant of Chromosome 15 Disrupting SMAD3 Reveals a Three-Generation Family at Serious Risk for Aortic Dissection. Clin. Genet. 2013, 83, 337–344. [Google Scholar] [CrossRef]
- Lee, S.-T.; Kim, J.-A.; Jang, S.-Y.; Kim, D.-K.; Kim, J.-W.; Ki, C.-S. A Novel COL3A1 Gene Mutation in Patient with Aortic Dissected Aneurysm and Cervical Artery Dissections. Heart Vessels 2008, 23, 144–148. [Google Scholar] [CrossRef]
- Koitabashi, N.; Yamaguchi, T.; Fukui, D.; Nakano, T.; Umeyama, A.; Toda, K.; Funada, R.; Ishikawa, M.; Kawamura, R.; Okada, K.; et al. Peripartum Iliac Arterial Aneurysm and Rupture in a Patient with Vascular Ehlers-Danlos Syndrome Diagnosed by Next-Generation Sequencing. Int. Heart J. 2018, 59, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrygiannis, G.; Loeys, B.; Defraigne, J.-O.; Sakalihasan, N. Cervical Artery Dissections and Type A Aortic Dissection in a Family with a Novel Missense COL3A1 Mutation of Vascular Type Ehlers–Danlos Syndrome. Eur. J. Med. Genet. 2015, 58, 634–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Yajima, J.; Oikawa, Y.; Ogasawara, K.; Uejima, T.; Abe, K.; Aizawa, T. Vascular Ehlers-Danlos Syndrome. J. Cardiol. 2009, 53, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, L.B.E.; Rolf, C.M.; Davis, G.J.; Hunsaker III, J.C. Sudden and Unexpected Death in Three Cases of Ehlers-Danlos Syndrome Type IV*: SUDDEN DEATH IN EHLERS-DANLOS TYPE IV. J. Forensic Sci. 2010, 55, 1641–1645. [Google Scholar] [CrossRef]
- Schwarze, U.; Goldstein, J.A.; Byers, P.H. Splicing Defects in the COL3A1 Gene: Marked Preference for 5′ (Donor) Spice-Site Mutations in Patients with Exon-Skipping Mutations and Ehlers-Danlos Syndrome Type IV. Am. J. Hum. Genet. 1997, 61, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Toda, M.; Kyoyama, H.; Nishimura, H.; Kojima, A.; Kuwabara, Y.; Kobayashi, Y.; Kikuchi, S.; Hirata, Y.; Moriyama, G.; et al. Vascular Ehlers-Danlos Syndrome with a Novel Missense Mutation in COL3A1: A Man in His 50s with Aortic Dissection after Interventional Treatment for Hemothorax as the First Manifestation. Intern. Med. Tokyo Jpn. 2019, 58, 3441–3447. [Google Scholar] [CrossRef] [Green Version]
- Shalhub, S.; Black, J.H.; Cecchi, A.C.; Xu, Z.; Griswold, B.F.; Safi, H.J.; Milewicz, D.M.; McDonnell, N.B. Molecular Diagnosis in Vascular Ehlers-Danlos Syndrome Predicts Pattern of Arterial Involvement and Outcomes. J. Vasc. Surg. 2014, 60, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Meienberg, J.; Rohrbach, M.; Neuenschwander, S.; Spanaus, K.; Giunta, C.; Alonso, S.; Arnold, E.; Henggeler, C.; Regenass, S.; Patrignani, A.; et al. Hemizygous Deletion of COL3A1, COL5A2, and MSTN Causes a Complex Phenotype with Aortic Dissection: A Lesson for and from True Haploinsufficiency. Eur. J. Hum. Genet. 2010, 18, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhuang, X.; Chen, B.; Wen, J.; Peng, F.; Liu, X.; Wei, M. 99-Case Study of Sporadic Aortic Dissection by Whole Exome Sequencing Indicated Novel Disease-Associated Genes and Variants in Chinese Population. BioMed Res. Int. 2020, 2020, 7857043. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, Y.; Li, Z.; Li, C.; Xiao, L.; Dai, J.; Li, S.; Liu, H.; Hu, D.; Wu, D.; et al. Identification of COL3A1 Variants Associated with Sporadic Thoracic Aortic Dissection: A Case-Control Study. Front. Med. 2021, 15, 438–447. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The Type of Variants at the COL3A1 Gene Associates with the Phenotype and Severity of Vascular Ehlers–Danlos Syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival Is Affected by Mutation Type and Molecular Mechanism in Vascular Ehlers-Danlos Syndrome (EDS Type IV). Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saratzis, A.; Bown, M.J. The Genetic Basis for Aortic Aneurysmal Disease. Heart Br. Card. Soc. 2014, 100, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokashi, S.A.; Svensson, L.G. Guidelines for the Management of Thoracic Aortic Disease in 2017. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 59–65. [Google Scholar] [CrossRef]
- Elefteriades, J.A. Thoracic Aortic Aneurysm: Reading the Enemy’s Playbook. Curr. Probl. Cardiol. 2008, 33, 203–277. [Google Scholar] [CrossRef]
- Rogers, A.M.; Hermann, L.K.; Booher, A.M.; Nienaber, C.A.; Williams, D.M.; Kazerooni, E.A.; Froehlich, J.B.; O’Gara, P.T.; Montgomery, D.G.; Cooper, J.V.; et al. Sensitivity of the Aortic Dissection Detection Risk Score, a Novel Guideline-Based Tool for Identification of Acute Aortic Dissection at Initial Presentation: Results from the International Registry of Acute Aortic Dissection. Circulation 2011, 123, 2213–2218. [Google Scholar] [CrossRef] [Green Version]
- Morello, F.; Piler, P.; Novak, M.; Kruzliak, P. Biomarkers for Diagnosis and Prognostic Stratification of Aortic Dissection: Challenges and Perspectives. Biomark. Med. 2014, 8, 931–941. [Google Scholar] [CrossRef]
- Nazerian, P.; Giachino, F.; Vanni, S.; Veglio, M.G.; Castelli, M.; Lison, D.; Bitossi, L.; Moiraghi, C.; Grifoni, S.; Morello, F. Diagnostic Performance of the Aortic Dissection Detection Risk Score in Patients with Suspected Acute Aortic Dissection. Eur. Heart J. Acute Cardiovasc. Care 2014, 3, 373–381. [Google Scholar] [CrossRef]
- Torbicki, A.; Perrier, A.; Konstantinides, S.; Agnelli, G.; Galiè, N.; Pruszczyk, P.; Bengel, F.; Brady, A.J.B.; Ferreira, D.; Janssens, U.; et al. Guidelines on the Diagnosis and Management of Acute Pulmonary Embolism: The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur. Heart J. 2008, 29, 2276–2315. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Högler, S.; Auer, J.; Berent, R.; Lassnig, E.; Kvas, E.; Eber, B. D-Dimer in Acute Aortic Dissection. Chest 2003, 123, 1375–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggebrecht, H.; Naber, C.K.; Bruch, C.; Kröger, K.; von Birgelen, C.; Schmermund, A.; Wichert, M.; Bartel, T.; Mann, K.; Erbel, R. Value of Plasma Fibrin D-Dimers for Detection of Acute Aortic Dissection. J. Am. Coll. Cardiol. 2004, 44, 804–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Distante, A.; Zizza, A.; Trimarchi, S.; Villani, M.; Salerno Uriarte, J.A.; De Luca Tupputi Schinosa, L.; Renzulli, A.; Sabino, F.; Nowak, R.; et al. Diagnosis of Acute Aortic Dissection by D-Dimer: The International Registry of Acute Aortic Dissection Substudy on Biomarkers (IRAD-Bio) Experience. Circulation 2009, 119, 2702–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimony, A.; Filion, K.B.; Mottillo, S.; Dourian, T.; Eisenberg, M.J. Meta-Analysis of Usefulness of d-Dimer to Diagnose Acute Aortic Dissection. Am. J. Cardiol. 2011, 107, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Newman, D.H. Evidence-Based Emergency Medicine. Can a Negative D-Dimer Result Rule out Acute Aortic Dissection? Ann. Emerg. Med. 2011, 58, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Schmoker, J.D.; McPartland, K.J.; Fellinger, E.K.; Boyum, J.; Trombley, L.; Ittleman, F.P.; Terrien, C.; Stanley, A.; Howard, A. Matrix Metalloproteinase and Tissue Inhibitor Expression in Atherosclerotic and Nonatherosclerotic Thoracic Aortic Aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 133, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Sinha, I.; Bethi, S.; Cronin, P.; Williams, D.M.; Roelofs, K.; Ailawadi, G.; Henke, P.K.; Eagleton, M.J.; Deeb, G.M.; Patel, H.J.; et al. A Biologic Basis for Asymmetric Growth in Descending Thoracic Aortic Aneurysms: A Role for Matrix Metalloproteinase 9 and 2. J. Vasc. Surg. 2006, 43, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Giachino, F.; Loiacono, M.; Lucchiari, M.; Manzo, M.; Battista, S.; Saglio, E.; Lupia, E.; Moiraghi, C.; Hirsch, E.; Mengozzi, G.; et al. Rule out of Acute Aortic Dissection with Plasma Matrix Metalloproteinase 8 in the Emergency Department. Crit. Care 2013, 17, R33. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Jiang, B.; Li, X.; Sun, H.; Li, X.; Jing, J.; Yang, J. Serum Matrix Metalloproteinase-9 Is a Valuable Biomarker for Identification of Abdominal and Thoracic Aortic Aneurysm: A Case-Control Study. BMC Cardiovasc. Disord. 2018, 18, 202. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xie, Y.; Liu, F.; Zhao, C.; Yu, R.; Ban, S.; Ye, Q.; Wen, J.; Wan, H.; Li, X.; et al. Expression of Matrix Metalloproteinase-12 in Aortic Dissection. BMC Cardiovasc. Disord. 2013, 13, 34. [Google Scholar] [CrossRef] [Green Version]
- Van Bogerijen, G.H.W.; Tolenaar, J.L.; Grassi, V.; Lomazzi, C.; Segreti, S.; Rampoldi, V.; Elefteriades, J.A.; Trimarchi, S. Biomarkers in TAA-the Holy Grail. Prog. Cardiovasc. Dis. 2013, 56, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.A.; Lindholt, J.S.; Hoskins, P.R.; Heickendorff, L.; Vammen, S.; Bradbury, A.W. The Relationship Between Abdominal Aortic Aneurysm Distensibility and Serum Markers of Elastin and Collagen Metabolism. Eur. J. Vasc. Endovasc. Surg. 2001, 21, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, T.; Suzuki, K.; Okada, M.; Shiigai, M.; Shimizu, M.; Maehara, T.; Ohsuzu, F. Soluble Elastin Fragments in Serum Are Elevated in Acute Aortic Dissection. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-M.; Shi, Y.-H.; Wang, J.-J.; Lü, F.-Q.; Gao, S. Elevated Plasma D-Dimer and Hypersensitive C-Reactive Protein Levels May Indicate Aortic Disorders. Rev. Bras. Cir. Cardiovasc. Orgao Off. Soc. Bras. Cir. Cardiovasc. 2011, 26, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, D.; Du, X.; Dong, J.-Z.; Zhou, X.-L.; Ma, C.-S. Value of D-Dimer and C Reactive Protein in Predicting Inhospital Death in Acute Aortic Dissection. Heart Br. Card. Soc. 2013, 99, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Zhou, X.-L.; Li, J.-J.; Luo, F.; Zhang, L.; Gao, L.-G.; Wang, L.-P.; Song, L.; Sun, K.; Zou, Y.-B.; et al. Plasma Concentrations of Interleukin-6, C-Reactive Protein, Tumor Necrosis Factor-α and Matrix Metalloproteinase-9 in Aortic Dissection. Clin. Chim. Acta Int. J. Clin. Chem. 2012, 413, 198–202. [Google Scholar] [CrossRef]
- Erdolu, B.; As, A.K. C-Reactive Protein and Neutrophil to Lymphocyte Ratio Values in Predicting Inhospital Death in Patients with Stanford Type A Acute Aortic Dissection. Heart Surg. Forum 2020, 23, E488–E492. [Google Scholar] [CrossRef]
- Sbarouni, E.; Georgiadou, P.; Analitis, A.; Chaidaroglou, A.; Marathias, A.; Degiannis, D.; Voudris, V. High Homocysteine and Low Folate Concentrations in Acute Aortic Dissection. Int. J. Cardiol. 2013, 168, 463–466. [Google Scholar] [CrossRef]
- Roohi, J.; Kang, B.; Bernard, D.; Bedja, D.; Dietz, H.C.; Brody, L.C. Moderately Elevated Homocysteine Does Not Contribute to Thoracic Aortic Aneurysm in Mice. J. Nutr. 2017, 147, 1290–1295. [Google Scholar] [CrossRef] [Green Version]
- Giusti, B.; Porciani, M.C.; Brunelli, T.; Evangelisti, L.; Fedi, S.; Gensini, G.F.; Abbate, R.; Sani, G.; Yacoub, M.; Pepe, G. Phenotypic Variability of Cardiovascular Manifestations in Marfan Syndrome. Possible Role of Hyperhomocysteinemia and C677T MTHFR Gene Polymorphism. Eur. Heart J. 2003, 24, 2038–2045. [Google Scholar] [CrossRef] [Green Version]
- Giusti, B.; Marcucci, R.; Lapini, I.; Sestini, I.; Lenti, M.; Yacoub, M.; Pepe, G. Role of Hyperhomocysteinemia in Aortic Disease. Cell. Mol. Biol. Noisy Gd. Fr. 2004, 50, 945–952. [Google Scholar]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-Beta Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Braverman, A.C. Transforming Growth Factor-β: A Biomarker in Marfan Syndrome? Circulation 2009, 120, 464–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stengl, R.; Ágg, B.; Pólos, M.; Mátyás, G.; Szabó, G.; Merkely, B.; Radovits, T.; Szabolcs, Z.; Benke, K. Potential Predictors of Severe Cardiovascular Involvement in Marfan Syndrome: The Emphasized Role of Genotype–Phenotype Correlations in Improving Risk Stratification—A Literature Review. Orphanet J. Rare Dis. 2021, 16, 245. [Google Scholar] [CrossRef]
- Matt, P.; Habashi, J.; Carrel, T.; Cameron, D.E.; Van Eyk, J.E.; Dietz, H.C. Recent Advances in Understanding Marfan Syndrome: Should We Now Treat Surgical Patients with Losartan? J. Thorac. Cardiovasc. Surg. 2008, 135, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; den Hartog, A.W.; de Waard, V.; Engele, L.; Radonic, T.; Lutter, R.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Zwinderman, A.H.; et al. Circulating Transforming Growth Factor-β as a Prognostic Biomarker in Marfan Syndrome. Int. J. Cardiol. 2013, 168, 2441–2446. [Google Scholar] [CrossRef]
- Ogawa, N.; Imai, Y.; Nishimura, H.; Kato, M.; Takeda, N.; Nawata, K.; Taketani, T.; Morota, T.; Takamoto, S.; Nagai, R.; et al. Circulating Transforming Growth Factor β-1 Level in Japanese Patients with Marfan Syndrome. Int. Heart. J. 2013, 54, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Forteza, A.; Evangelista, A.; Sánchez, V.; Teixidó, G.; García, D.; Sanz, P.; Gutiérrez, L.; Centeno, J.; Rodríguez-Palomares, J.; Cortina, J.; et al. Valoración de la eficacia y la seguridad del losartán frente al atenolol en la prevención de la dilatación de la aorta en el síndrome de Marfan. Rev. Esp. Cardiol. 2011, 64, 492–498. [Google Scholar] [CrossRef]
- Suzuki, T.; Trimarchi, S.; Sawaki, D.; Grassi, V.; Costa, E.; Rampoldi, V.; Nagai, R.; Eagle, K. Circulating Transforming Growth Factor-Beta Levels in Acute Aortic Dissection. J. Am. Coll. Cardiol. 2011, 58, 775. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.-M.; Lin, H. Expressions of Transforming Growth Factor Β1 Signaling Cytokines in Aortic Dissection. Braz. J. Cardiovasc. Surg. 2018, 33, 597–602. [Google Scholar] [CrossRef]
- Tellides, G. Further Evidence Supporting a Protective Role of Transforming Growth Factor-β (TGFβ) in Aortic Aneurysm and Dissection. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1983–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, H.; Takeda, N.; Fujiwara, T.; Yagi, H.; Maemura, S.; Kanaya, T.; Nawata, K.; Morita, H.; Komuro, I. Activation of TGF-β Signaling in an Aortic Aneurysm in a Patient with Loeys-Dietz Syndrome Caused by a Novel Loss-of-Function Variant of TGFBR1. Hum. Genome Var. 2019, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.A.; Chakrabarti, M.; Ferruzzi, J.; Azhar, M.; Eberth, J.F. Mechanics of Ascending Aortas from TGFβ-1, -2, -3 Haploinsufficient Mice and Elastase-Induced Aortopathy. J. Biomech. 2021, 125, 110543. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, M.; Millot, N.; Sheikhzadeh, S.; Rybczynski, M.; Gerth, S.; Kölbel, T.; Keyser, B.; Kutsche, K.; Robinson, P.N.; Berger, J.; et al. Total Serum Transforming Growth Factor-Β1 Is Elevated in the Entire Spectrum of Genetic Aortic Syndromes: TGF-Β1 Serum Levels in GAS. Clin. Cardiol. 2014, 37, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, H.; Suzuki, T.; Hiroi, Y.; Ohtaki, E.; Suzuki, S.; Yazaki, Y.; Nagai, R. Diagnosis of Aortic Dissection by Immunoassay for Circulating Smooth Muscle Myosin. Lancet 1995, 345, 191–192. [Google Scholar] [CrossRef]
- Suzuki, T.; Katoh, H.; Watanabe, M.; Kurabayashi, M.; Hiramori, K.; Hori, S.; Nobuyoshi, M.; Tanaka, H.; Kodama, K.; Sato, H.; et al. Novel Biochemical Diagnostic Method for Aortic Dissection: Results of a Prospective Study Using an Immunoassay of Smooth Muscle Myosin Heavy Chain. Circulation 1996, 93, 1244–1249. [Google Scholar] [CrossRef]
- Suzuki, T.; Katoh, H.; Tsuchio, Y.; Hasegawa, A.; Kurabayashi, M.; Ohira, A.; Hiramori, K.; Sakomura, Y.; Kasanuki, H.; Hori, S.; et al. Diagnostic Implications of Elevated Levels of Smooth-Muscle Myosin Heavy-Chain Protein in Acute Aortic Dissection. The Smooth Muscle Myosin Heavy Chain Study. Ann. Intern. Med. 2000, 133, 537–541. [Google Scholar] [CrossRef]
- Davidson, E.; Weinberger, I.; Rotenberg, Z.; Fuchs, J.; Maler, S.; Agmon, J. Elevated Serum Creatine Kinase Levels. An Early Diagnostic Sign of Acute Dissection of the Aorta. Arch. Intern. Med. 1988, 148, 2184–2186. [Google Scholar] [CrossRef]
- Suzuki, T.; Katoh, H.; Kurabayashi, M.; Yazaki, Y.; Nagai, R. Biochemical Diagnosis of Aortic Dissection by Raised Concentrations of Creatine Kinase BB-Isozyme. Lancet 1997, 350, 784–785. [Google Scholar] [CrossRef]
- Suzuki, T.; Distante, A.; Zizza, A.; Trimarchi, S.; Villani, M.; Salerno Uriarte, J.A.; de Luca Tupputi Schinosa, L.; Renzulli, A.; Sabino, F.; Nowak, R.; et al. Preliminary Experience with the Smooth Muscle Troponin-like Protein, Calponin, as a Novel Biomarker for Diagnosing Acute Aortic Dissection. Eur. Heart J. 2008, 29, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Peng, Z.; Chai, X.; Zhu, Q.; Yang, G.; Zhao, Q.; Zhou, S. Potential Biomarkers for Early Diagnosis of Acute Aortic Dissection. Heart Lung J. Crit. Care 2015, 44, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qian, H.; Yang, Q.; Hu, J.; Gan, C.; Meng, W. Relationship between the Extent of Dissection and Platelet Activation in Acute Aortic Dissection. J. Cardiothorac. Surg. 2015, 10, 162. [Google Scholar] [CrossRef] [Green Version]
- Sbarouni, E.; Georgiadou, P.; Analitis, A.; Voudris, V. Significant Changes in Platelet Count, Volume and Size in Acute Aortic Dissection. Int. J. Cardiol. 2013, 168, 4349–4350. [Google Scholar] [CrossRef]
- Huang, B.; Tian, L.; Fan, X.; Zhu, J.; Liang, Y.; Yang, Y. Low Admission Platelet Counts Predicts Increased Risk of In-Hospital Mortality in Patients with Type A Acute Aortic Dissection. Int. J. Cardiol. 2014, 172, e484–e486. [Google Scholar] [CrossRef] [PubMed]
- König, K.C.; Lahm, H.; Dreßen, M.; Doppler, S.A.; Eichhorn, S.; Beck, N.; Kraehschuetz, K.; Doll, S.; Holdenrieder, S.; Kastrati, A.; et al. Aggrecan: A New Biomarker for Acute Type A Aortic Dissection. Sci. Rep. 2021, 11, 10371. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, X.-W.; Lu, W.-H.; Cheng, J.; Wu, X.-Y.; Ai, R.; Zhou, Z.-H.; Tang, Z.-Z.; Liao, Y.-H. High-Sensitivity Cardiac Troponin T: A Biomarker for the Early Risk Stratification of Type-A Acute Aortic Dissection? Arch. Cardiovasc. Dis. 2016, 109, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiao, X.; Li, L.; Hu, C.; Zhang, X.; Pan, L.; Yu, H.; Li, J.; Chen, D.; Du, J.; et al. Increased Circulating Angiopoietin-Like Protein 8 Levels Are Associated with Thoracic Aortic Dissection and Higher Inflammatory Conditions. Cardiovasc. Drugs Ther. 2020, 34, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Jerves, T.; Beaton, A.; Kruszka, P. The Genetic Workup for Structural Congenital Heart Disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 178–186. [Google Scholar] [CrossRef]
- Rigelsky, C.M.; Moran, R.T. Genetics of Syndromic and Nonsyndromic Aortopathies. Curr. Opin. Pediatr. 2019, 31, 694–701. [Google Scholar] [CrossRef]
- SEC Working Group for ESC 2014 Guidelines on Diagnosis and Treatment of Aortic Diseases; Expert Reviewers for ESC 2014 Guidelines on Diagnosis and Treatment of Aortic Diseases; SEC Clinical Practice Guidelines Committee. Comments on the 2014 ESC Guidelines on the Diagnosis and Treatment of Aortic Diseases. Rev. Esp. Cardiol. Engl. Ed. 2015, 68, 179–184. [Google Scholar] [CrossRef]
- Writing Group Members; Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients with Thoracic Aortic Disease: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.-C.; Hostetler, E.M.; Fan, Y.; Kulmacz, R.J.; Zhang, D.; GenTAC Investigators; Nickerson, D.A.; Leal, S.M.; LeMaire, S.A.; Regalado, E.S.; et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J. Am. Coll. Cardiol. 2017, 70, 2728–2730. [Google Scholar] [CrossRef] [PubMed]
- Hicks, K.L.; Byers, P.H.; Quiroga, E.; Pepin, M.G.; Shalhub, S. Testing Patterns for Genetically Triggered Aortic and Arterial Aneurysms and Dissections at an Academic Center. J. Vasc. Surg. 2018, 68, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, T.; Tröger, H.D.; Schmidtke, J. The Genetic Message of a Sudden, Unexpected Death Due to Thoracic Aortic Dissection. Forensic Sci. Int. 2009, 187, 1–5. [Google Scholar] [CrossRef]
- Boileau, A.; Lindsay, M.; Michel, J.-B.; Devaux, Y. Epigenetics in Ascending Thoracic Aortic Aneurysm and Dissection. AORTA 2018, 06, 001–012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Barbacioru, C.C.; Shiffman, D.; Balasubramanian, S.; Iakoubova, O.; Tranquilli, M.; Albornoz, G.; Blake, J.; Mehmet, N.N.; Ngadimo, D.; et al. Gene Expression Signature in Peripheral Blood Detects Thoracic Aortic Aneurysm. PLoS ONE 2007, 2, e1050. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.G.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in Aortic Dilation: Implications for Aneurysm Formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Merk, D.R.; Chin, J.T.; Dake, B.A.; Maegdefessel, L.; Miller, M.O.; Kimura, N.; Tsao, P.S.; Iosef, C.; Berry, G.J.; Mohr, F.W.; et al. MiR-29b Participates in Early Aneurysm Development in Marfan Syndrome. Circ. Res. 2012, 110, 312–324. [Google Scholar] [CrossRef]
- Van Rooij, E. The Art of MicroRNA Research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.G.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The Knockout of MiR-143 and -145 Alters Smooth Muscle Cell Maintenance and Vascular Homeostasis in Mice: Correlates with Human Disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.-Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a Novel Smooth Muscle Cell Phenotypic Marker and Modulator, Controls Vascular Neointimal Lesion Formation. Circ. Res. 2009, 105, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Stroud, R.E.; O’Quinn, E.C.; Black, L.E.; Barth, J.L.; Elefteriades, J.A.; Bavaria, J.E.; Gorman, J.H.; Gorman, R.C.; Spinale, F.G.; et al. Selective MicroRNA Suppression in Human Thoracic Aneurysms: Relationship of MiR-29a to Aortic Size and Proteolytic Induction. Circ. Cardiovasc. Genet. 2011, 4, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, P.; Phillippi, J.; Chukkapalli, S.; Rivera-Kweh, M.; Velsko, I.; Gleason, T.; VanRyzin, P.; Aalaei-Andabili, S.; Ghanta, R.; Beaver, T.; et al. Aneurysm-Specific MiR-221 and MiR-146a Participates in Human Thoracic and Abdominal Aortic Aneurysms. Int. J. Mol. Sci. 2017, 18, 875. [Google Scholar] [CrossRef]
- Patuzzo, C.; Pasquali, A.; Trabetti, E.; Malerba, G.; Pignatii, P.; Tessari, M.; Faggian, G. A Preliminary MicroRNA Analysis of Non Syndromic Thoracic Aortic Aneurysms. Balk. J. Med. Genet. 2012, 15, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Pei, H.; Tian, C.; Sun, X.; Qian, X.; Liu, P.; Liu, W.; Chang, Q. Overexpression of MicroRNA-145 Promotes Ascending Aortic Aneurysm Media Remodeling through TGF-Β1. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2015, 49, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, M.; Zou, S.; Weng, J.; Hou, L.; Yang, L.; Zhao, Z.; Bao, J.; Jing, Z. A MicroRNA Profile Comparison between Thoracic Aortic Dissection and Normal Thoracic Aorta Indicates the Potential Role of MicroRNAs in Contributing to Thoracic Aortic Dissection Pathogenesis. J. Vasc. Surg. 2011, 53, 1341–1349.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Huang, H.; Sun, X.; Guo, Y.; Hamblin, M.; Ritchie, R.P.; Garcia-Barrio, M.T.; Zhang, J.; Chen, Y.E. MicroRNA-1 Regulates Smooth Muscle Cell Differentiation by Repressing Kruppel-Like Factor 4. Stem Cells Dev. 2011, 20, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 Controls Vascular Smooth Muscle Cell Phenotypic Switch in Vitro and Vascular Remodeling In Vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Liu, Y.; Yi, B.; Wang, G.; You, X.; Zhao, X.; Summer, R.; Qin, Y.; Sun, J. MicroRNA-638 Is Highly Expressed in Human Vascular Smooth Muscle Cells and Inhibits PDGF-BB-Induced Cell Proliferation and Migration through Targeting Orphan Nuclear Receptor NOR1. Cardiovasc. Res. 2013, 99, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Merlet, E.; Atassi, F.; Motiani, R.K.; Mougenot, N.; Jacquet, A.; Nadaud, S.; Capiod, T.; Trebak, M.; Lompré, A.-M.; Marchand, A. MiR-424/322 Regulates Vascular Smooth Muscle Cell Phenotype and Neointimal Formation in the Rat. Cardiovasc. Res. 2013, 98, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-S.; Wang, H.-Y.J.; Liao, Y.-C.; Tsai, P.-C.; Chen, K.-C.; Cheng, H.-Y.; Lin, R.-T.; Juo, S.-H.H. MicroRNA-195 Regulates Vascular Smooth Muscle Cell Phenotype and Prevents Neointimal Formation. Cardiovasc. Res. 2012, 95, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-Specific Effects of MiR-221/222 in Vessels: Molecular Mechanism and Therapeutic Application. J. Mol. Cell. Cardiol. 2012, 52, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a Is a Novel Regulator of Vascular Smooth Muscle Cell Function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Xiong, W.; Yuan, J.; Li, J.; Liu, J.; Xu, X. MiRNA-146a Regulates the Maturation and Differentiation of Vascular Smooth Muscle Cells by Targeting NF-ΚB Expression. Mol. Med. Rep. 2013, 8, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, F.; Yu, X.; Lu, X.; Zheng, G. MicroRNA-155 Modulates the Proliferation of Vascular Smooth Muscle Cells by Targeting Endothelial Nitric Oxide Synthase. Int. J. Mol. Med. 2015, 35, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cheng, Y.; Chen, X.; Yang, J.; Xu, L.; Zhang, C. MicroRNA-31 Regulated by the Extracellular Regulated Kinase Is Involved in Vascular Smooth Muscle Cell Growth via Large Tumor Suppressor Homolog 2. J. Biol. Chem. 2011, 286, 42371–42380. [Google Scholar] [CrossRef] [Green Version]
- Li, T.-J.; Chen, Y.-L.; Gua, C.-J.; Xue, S.-J.; Ma, S.-M.; Li, X.-D. MicroRNA 181b Promotes Vascular Smooth Muscle Cells Proliferation through Activation of PI3K and MAPK Pathways. Int. J. Clin. Exp. Pathol. 2015, 8, 10375–10384. [Google Scholar]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA Expression Signature and Antisense-Mediated Depletion Reveal an Essential Role of MicroRNA in Vascular Neointimal Lesion Formation. Circ. Res. 2007, 100, 1579–1588. [Google Scholar] [CrossRef]
- Yang, J.; Chen, L.; Ding, J.; Fan, Z.; Li, S.; Wu, H.; Zhang, J.; Yang, C.; Wang, H.; Zeng, P.; et al. MicroRNA-24 Inhibits High Glucose-Induced Vascular Smooth Muscle Cell Proliferation and Migration by Targeting HMGB1. Gene 2016, 586, 268–273. [Google Scholar] [CrossRef]
- Moushi, A.; Michailidou, K.; Soteriou, M.; Cariolou, M.; Bashiardes, E. MicroRNAs as Possible Biomarkers for Screening of Aortic Aneurysms: A Systematic Review and Validation Study. Biomarkers 2018, 23, 253–264. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, F.; Doldo, E.; Pisano, C.; Scioli, M.G.; Centofanti, F.; Proietti, G.; Falconi, M.; Sangiuolo, F.; Ferlosio, A.; Ruvolo, G.; et al. Specific MiRNA and Gene Deregulation Characterize the Increased Angiogenic Remodeling of Thoracic Aneurysmatic Aortopathy in Marfan Syndrome. Int. J. Mol. Sci. 2020, 21, 6886. [Google Scholar] [CrossRef] [PubMed]
- Patamsytė, V.; Žukovas, G.; Gečys, D.; Žaliaduonytė, D.; Jakuška, P.; Benetis, R.; Lesauskaitė, V. Long Noncoding RNAs CARMN, LUCAT1, SMILR, and MALAT1 in Thoracic Aortic Aneurysm: Validation of Biomarkers in Clinical Samples. Dis. Markers 2020, 2020, 8521899. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, G.; Jing, Y.; He, X.; Dong, J.; Zheng, J.; Zou, M.; Li, H.; Wang, S.; Sun, Y.; et al. LncRNA Expression Profile of Human Thoracic Aortic Dissection by High-Throughput Sequencing. Cell. Physiol. Biochem. 2018, 46, 1027–1041. [Google Scholar] [CrossRef]
- Xiao, W.; Li, X.; Ji, C.; Shi, J.; Pan, Y. LncRNA Sox2ot Modulates the Progression of Thoracic Aortic Aneurysm by Regulating MiR-330-5p/Myh11. Biosci. Rep. 2020, 40, BSR20194040. [Google Scholar] [CrossRef]
- Zhao, X.; Cheng, S.; Li, S.; Li, J.; Bai, X.; Xi, J. CDKN2B-AS1 Aggravates the Pathogenesis of Human Thoracic Aortic Dissection by Sponge to MiR-320d. J. Cardiovasc. Pharmacol. 2020, 76, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Cui, M.; Fu, X.; Ma, J.; Zhang, K.; Zhang, D.; Zhai, S. LncRNA Xist Induces Arterial Smooth Muscle Cell Apoptosis in Thoracic Aortic Aneurysm through MiR-29b-3p/Eln Pathway. Biomed. Pharmacother. 2021, 137, 111163. [Google Scholar] [CrossRef]
- Ren, M.; Wang, T.; Wei, X.; Wang, Y.; Ouyang, C.; Xie, Y.; Ye, X.; Han, Z. LncRNA H19 Regulates Smooth Muscle Cell Functions and Participates in the Development of Aortic Dissection through Sponging MiR-193b-3p. Biosci. Rep. 2021, 41, BSR20202298. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Zhang, M.; Wu, Q.; Shi, F. Lnc-OIP5-AS1 Exacerbates Aorta Wall Injury during the Development of Aortic Dissection through Upregulating TUB via Sponging MiR-143-3p. Life Sci. 2021, 271, 119199. [Google Scholar] [CrossRef]
- Boeckel, J.-N.; Jaé, N.; Heumüller, A.W.; Chen, W.; Boon, R.A.; Stellos, K.; Zeiher, A.M.; John, D.; Uchida, S.; Dimmeler, S. Identification and Characterization of Hypoxia-Regulated Endothelial Circular RNA. Circ. Res. 2015, 117, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Tang, X.; Zhu, X.; Zhou, Q.; Guo, Y.; Zhao, R.; Wang, D.; Gong, B. Expression Profiles of CircRNAs and the Potential Diagnostic Value of Serum CircMARK3 in Human Acute Stanford Type A Aortic Dissection. PLoS ONE 2019, 14, e0219013. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Yang, Y.; Xin, H.; Li, M.; Zong, T.; He, X.; Yu, T.; Xin, H. Non-Coding RNAs in Aortic Dissection: From Biomarkers to Therapeutic Targets. J. Cell. Mol. Med. 2020, 24, 11622–11637. [Google Scholar] [CrossRef] [PubMed]
- Gago-Díaz, M.; Blanco-Verea, A.; Teixidó-Turà, G.; Valenzuela, I.; Del Campo, M.; Borregan, M.; Sobrino, B.; Amigo, J.; García-Dorado, D.; Evangelista, A.; et al. Whole Exome Sequencing for the Identification of a New Mutation in TGFB2 Involved in a Familial Case of Non-Syndromic Aortic Disease. Clin. Chim. Acta 2014, 437, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Regalado, E.S.; Shendure, J.; Nickerson, D.A.; Guo, D. Successes and Challenges of Using Whole Exome Sequencing to Identify Novel Genes Underlying an Inherited Predisposition for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Trends Cardiovasc. Med. 2014, 24, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Zhang, W.; Liu, C.; Zhou, M.; Ran, F.; Yi, L.; Sun, X.; Liu, Z. Whole Exome Sequencing Identifies FBN1 Mutations in Two Patients with Early-onset Type B Aortic Dissection. Mol. Med. Rep. 2017, 16, 6620–6625. [Google Scholar] [CrossRef] [Green Version]
- Regalado, E.S.; Guo, D.C.; Santos-Cortez, R.L.P.; Hostetler, E.; Bensend, T.A.; Pannu, H.; Estrera, A.; Safi, H.; Mitchell, A.L.; Evans, J.P.; et al. Pathogenic FBN1 Variants in Familial Thoracic Aortic Aneurysms and Dissections. Clin. Genet. 2016, 89, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Landis, B.J.; Schubert, J.A.; Lai, D.; Jegga, A.G.; Shikany, A.R.; Foroud, T.; Ware, S.M.; Hinton, R.B. Exome Sequencing Identifies Candidate Genetic Modifiers of Syndromic and Familial Thoracic Aortic Aneurysm Severity. J Cardiovasc. Transl. Res. 2017, 10, 423–432. [Google Scholar] [CrossRef]
- Li, Y.; Gao, S.; Han, Y.; Song, L.; Kong, Y.; Jiao, Y.; Huang, S.; Du, J.; Li, Y. Variants of Focal Adhesion Scaffold Genes Cause Thoracic Aortic Aneurysm. Circ. Res. 2021, 128, 8–23. [Google Scholar] [CrossRef]
- Li, Y.; Fang, M.; Yang, J.; Yu, C.; Kuang, J.; Sun, T.; Fan, R. Analysis of the Contribution of 129 Candidate Genes to Thoracic Aortic Aneurysm or Dissection of a Mixed Cohort of Sporadic and Familial Cases in South China. Am. J. Transl. Res. 2021, 13, 4281–4295. [Google Scholar]
- Erhart, P.; Brandt, T.; Straub, B.K.; Hausser, I.; Hentze, S.; Böckler, D.; Grond-Ginsbach, C. Familial Aortic Disease and a Large Duplication in Chromosome 16p13.1. Mol. Genet. Genomic Med. 2018, 6, 441–445. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Li, J.; Wang, Y.; Cheng, Y.; Wang, C.; Shu, X. Recurrent Germline Mutations as Genetic Markers for Aortic Root Dilatation in Bicuspid Aortic Valve Patients. Heart Vessels 2021, 36, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Overwater, E.; Marsili, L.; Baars, M.J.H.; Baas, A.F.; van de Beek, I.; Dulfer, E.; van Hagen, J.M.; Hilhorst-Hofstee, Y.; Kempers, M.; Krapels, I.P.; et al. Results of Next-Generation Sequencing Gene Panel Diagnostics Including Copy-Number Variation Analysis in 810 Patients Suspected of Heritable Thoracic Aortic Disorders. Hum. Mutat. 2018, 39, 1173–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Hu, M.; Fedak, P.W.M.; Oudit, G.Y.; Kassiri, Z. Cell-Specific Functions of ADAM17 Regulate the Progression of Thoracic Aortic Aneurysm. Circ. Res. 2018, 123, 372–388. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Gao, S.; Zhou, M.; Qi, F.; Ding, N.; Zhang, J.; Li, R.; Wang, J.; Shi, J.; et al. Excessive DNA Damage Mediates ECM Degradation via the RBBP8/NOTCH1 Pathway in Sporadic Aortic Dissection. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2022, 1868, 166303. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Lim, Z.; Dean, P.H.; Potts, J.E.; Tang, J.N.C.; Etheridge, S.P.; Lara, A.; Husband, P.; Sherwin, E.D.; Ackerman, M.J.; et al. Genetic Insurance Discrimination in Sudden Arrhythmia Death Syndromes: Empirical Evidence from a Cross-Sectional Survey in North America. Circ. Cardiovasc. Genet. 2017, 10, e001442. [Google Scholar] [CrossRef] [Green Version]
- Mehta, C.K.; Son, A.Y.; Chia, M.C.; Budd, A.N.; Allen, B.D.; Vassallo, P.; Hoel, A.W.; Brady, W.J.; Nable, J.V. Management of Acute Aortic Syndromes from Initial Presentation to Definitive Treatment. Am. J. Emerg. Med. 2022, 51, 108–113. [Google Scholar] [CrossRef]



| Biological Process/Cellular Compartment | Gene | Protein | OMIM | Syndromic TAA/D | Non-Syndromic FTAA/D | Associated Syndrome/Diseases |
|---|---|---|---|---|---|---|
| Extracellular matrix/remodeling | BGN | Biglycan | 300,989 | + | − | Meester-Loeys syndrome. ARD, TAAD, pulmonary artery aneurysm, IA, arterial tortuosity [19]. |
| COL3A1 | Collagen Type III α1 Chain | 130,050 | + | − | EDS, vascular type IV. TAAD, early aortic dissection, visceral arterial dissection, vessel fragility [20]. | |
| EFEMP2 | EGF Containing Fibulin Extracellular Matrix Protein 2 | 614,437 | + | − | Cutis laxa, AR type Ib. Ascending aortic aneurysms, other arterial aneurysms, arterial tortuosity, stenosis [21]. | |
| ELN | Elastin | 123,700 185,500 | + | − | Cutis laxa. AD ARD, ascending aortic aneurysm and dissection [22], TAA [23,24], BAV, IA possibly associated with SVAS. | |
| FBN1 | Fibrillin-1 | 154,700 | + | + | Marfan syndrome. ARD, TAA [25], TAAD [26], AAA, other arterial aneurysms, pulmonary artery dilatation, arterial tortuosity [27]. | |
| LOX | Protein-lysine 6-oxidase | 617,168 | − | + | AAT10. AAA, hepatic artery aneurysm, BAV, CAD, TAAD [28,29]. | |
| MFAP5 | Microfibril Associated Protein 5 | 616,166 | − | + | AAT9. ARD, TAA [30,31]. | |
| Smooth muscle cells | ACTA2 | Smooth muscle α-actin | 611,788 613,834 614,042 | + | + | AAT6, multisystemic smooth muscle dysfunction, MYMY5. Early aortic dissection, CAD, stroke (moyamoya disease), PDA, pulmonary artery dilation, BAV, TAAD, TAA [24,32]. |
| FLNA | Filamin A | 300,049 | + | − | Periventricular nodular heterotopia and otopalatodigital syndrome. Aortic dilatation/aneurysms, peripheral arterial dilatation, PDA, IA, BAV, TAA [32,33]. | |
| MYH11 | Smooth muscle myosin heavy chain | 132,900 | − | + | AAT4. PDA, CAD, peripheral vascular occlusive disease, carotid IA, TAAD, early aortic dissection [32,34,35]. | |
| MYLK | Myosin light chain kinase | 613,780 | − | + | AAT7. TAAD, early aortic dissections [36,37]. | |
| TGF-β signaling | LTBP1 | Latent TGF-β binding protein 1 | 150,390 | + | − | Aortic dilation with associated musculoskeletal findings. Dental anomalies, short stature. TAAD, AAA, visceral and peripheral arterial aneurysm [38]. |
| LTBP3 | Latent TGF-β binding protein 3 | 602,090 | ||||
| SMAD2 | SMAD2 | 619,657 619,656 | + | - | Unidentified CTD with arterial aneurysm/dissections. ARD, ascending aortic aneurysms, vertebral/carotid aneurysms and dissections [39], AAA. | |
| SMAD3 | SMAD3 | 613,795 | + | + | LDS type III. ARD, TAAD [40], early aortic dissection [39], AAA, arterial tortuosity, other arterial aneurysms/dissections [9], IA, BAV. | |
| SMAD4 | SMAD4 | 175,050 | + | - | JP/HHT syndrome. ARD, TAAD [39], AVMs, IA. | |
| SMAD6 | SMAD6 | 602,931 | - | + | AOVD2. BAV/TAA [24]. | |
| TGFB2 | TGF-β2 | 614,816 | + | + | LDS type IV. ARD, TAA [40], TAAD, arterial tortuosity [39], other arterial aneurysms, BAV. | |
| TGFB3 | TGF-β3 | 615,582 | + | - | LDS type V. ARD, TAAD, AAA/dissection, other arterial aneurysms, IA/dissection [39]. | |
| TGFBR1 | TGF-β receptor type 1 | 609,192 | + | + | LDS type I+AAT5. TAAD [40], early aortic dissection, AAA, arterial tortuosity, other arterial aneurysms/dissection [9], IA, PDA, BAV. | |
| TGFBR2 | TGF-β receptor type 2 | 610,168 | + | + | LDS type II+AAT3. TAAD [40], early aortic dissection, AAA, arterial tortuosity, other arterial aneurysms/dissection [9], IA, PDA, BAV. | |
| Others | AXIN1/PDIA2 locus | − | − | + | − | BAV. BAV/TAA [41]. |
| FBN2 | Fibrillin-2 | 121,050 | + | − | Contractual arachnodactyly. Rare ARD and aortic dissection [42], BAV, PDA. | |
| FOXE3 | Forkhead box 3 | 617,349 | − | + | AAT11. TAAD [30] (primarily type A dissection). | |
| MAT2A | Methionine adenosyl-transferase II α | n.a. | − | + | FTAA Thoracic aortic aneurysms [30,43]. BAV. | |
| NOTCH1 | NOTCH1 | 109,730 | − | + | AOVD1. BAV/TAAD [24]. | |
| PRKG1 | Type 1 cGMP-dependent protein kinase | 615,436 | − | + | AAT8. TAAD [28,43], early aortic dissection, AAA, coronary artery aneurysm/dissection, aortic tortuosity, small vessel, CVD. | |
| ROBO4 | Roundabout guidance receptor 4 | 607,528 | − | + | BAV. BAV/TAA [24]. | |
| SKI | Sloan Kettering proto-oncoprotein | 182,212 | + | − | Shprintzen–Goldberg syndrome. ARD, arterial tortuosity, pulmonary artery dilation, other (splenic) arterial aneurysms [36]. | |
| SLC2A10 | Glucose transporter 10 | 208,050 | + | − | Arterial tortuosity syndrome. ARD, ascending aortic aneurysms [36], other arterial aneurysms, arterial tortuosity [44], elongated arteries, aortic/pulmonary artery stenosis. |
| Marker | Animal Models | Human Cohort | TAA | TAAD |
|---|---|---|---|---|
| ANGPTL8 | - | 78 patients with AD and 72 controls [170] | ||
| Calponin | - | 217 patients with AD [163] | + | + |
| CK-BB | - | 10 patients with AAD [162] | ||
| CK-MM | - | 22 patients with AAD [161] | ||
| CRP | - | 49 patients with aortic disorders [130] | + | + |
| - | 114 patients with AAD [139] | |||
| - | 118 patients with AAD [140] | |||
| CSPCP (aggrecan) | - | 33 patients with AAD [168] | + | + |
| cTnT | - | 103 patients with AAD [169] | ||
| DD | - | 24 patients with AD/TAAD [124] | + | |
| - | 64 patients with AD [125] | |||
| - | 220 patients with AAD [126] | |||
| Hcy | - | 31 patients with AAD [141] | + | |
| C57BL/6J mice [142] | - | |||
| MMP8 | - | 186 patients suspected AAD [131] | ||
| MMP9 | - | 105 patients with AAA, 79 with TAA, 112 controls [132] | + | |
| MMP12 | - | 15 patients with AAD, 10 controls [133] | ||
| MPV/PLT | - | 300 patients with aortic disorders [166] | + | |
| - | 183 patients with AAD [167] | |||
| sELAFs | - | 62 patients with AAA [135] | ||
| - | 25 patients with AAD [136] | |||
| smMHC | Mice [158] | |||
| - | 27 patients with AD [159] | + | ||
| TIMP1 | - | 93 patients with TAA and 24 controls [70] | + | |
| TIMP2 | - | 93 patients with TAA and 24 controls [70] | + | |
| TGF-β | - | 50 families with LDS [145] | + | |
| - | 28 patients with AAD [152] | |||
| - | 40 patients with aortic disorders [153] | + | + | |
| - | 1 patient with LDS [155] | + |
| Study/Methodology | Genes Identified | Animal Models | Human Cohort |
|---|---|---|---|
| WES (WHOLE EXOME SEQUENCING) | MLX, DAB2IP, EP300, ZFYVE9, PML, PRKCD | - | 99 patients with TAA [112] |
| ADCK4, COL15A1 | - | 27 patients with fTAA [220] | |
| TES, TLN1, ZYX | C57/BL6 mice | 556 patients with sporadic TAA and 1092 controls [221] | |
| MCTP2 | - | 151 patients with TAAD [222] | |
| 16p13.1 duplication | - | 1 patient with fTAAD [223] | |
| C1R | - | 13 patients with BAV [224] | |
| NGS PANELS | SCARF2 | - | 810 cases of suspected TAA [225] |
| MOUSE MODELS | ADAM17 | Sm22α-Cre mice [226] | - |
| RBBP8 | Male C57/BL6 mice | 12 Aortic aneurysm/dissection samples [227] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cario, R.; Giannini, M.; Cassioli, G.; Kura, A.; Gori, A.M.; Marcucci, R.; Nistri, S.; Pepe, G.; Giusti, B.; Sticchi, E. Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection. Diagnostics 2022, 12, 1785. https://doi.org/10.3390/diagnostics12081785
De Cario R, Giannini M, Cassioli G, Kura A, Gori AM, Marcucci R, Nistri S, Pepe G, Giusti B, Sticchi E. Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection. Diagnostics. 2022; 12(8):1785. https://doi.org/10.3390/diagnostics12081785
Chicago/Turabian StyleDe Cario, Rosina, Marco Giannini, Giulia Cassioli, Ada Kura, Anna Maria Gori, Rossella Marcucci, Stefano Nistri, Guglielmina Pepe, Betti Giusti, and Elena Sticchi. 2022. "Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection" Diagnostics 12, no. 8: 1785. https://doi.org/10.3390/diagnostics12081785
APA StyleDe Cario, R., Giannini, M., Cassioli, G., Kura, A., Gori, A. M., Marcucci, R., Nistri, S., Pepe, G., Giusti, B., & Sticchi, E. (2022). Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection. Diagnostics, 12(8), 1785. https://doi.org/10.3390/diagnostics12081785