
Crafting a Personalized Prognostic Model for Malignant Prostate Cancer Patients Using Risk Gene Signatures Discovered through TCGA-PRAD Mining, Machine Learning, and Single-Cell RNA-Sequencing
 , ,
, , 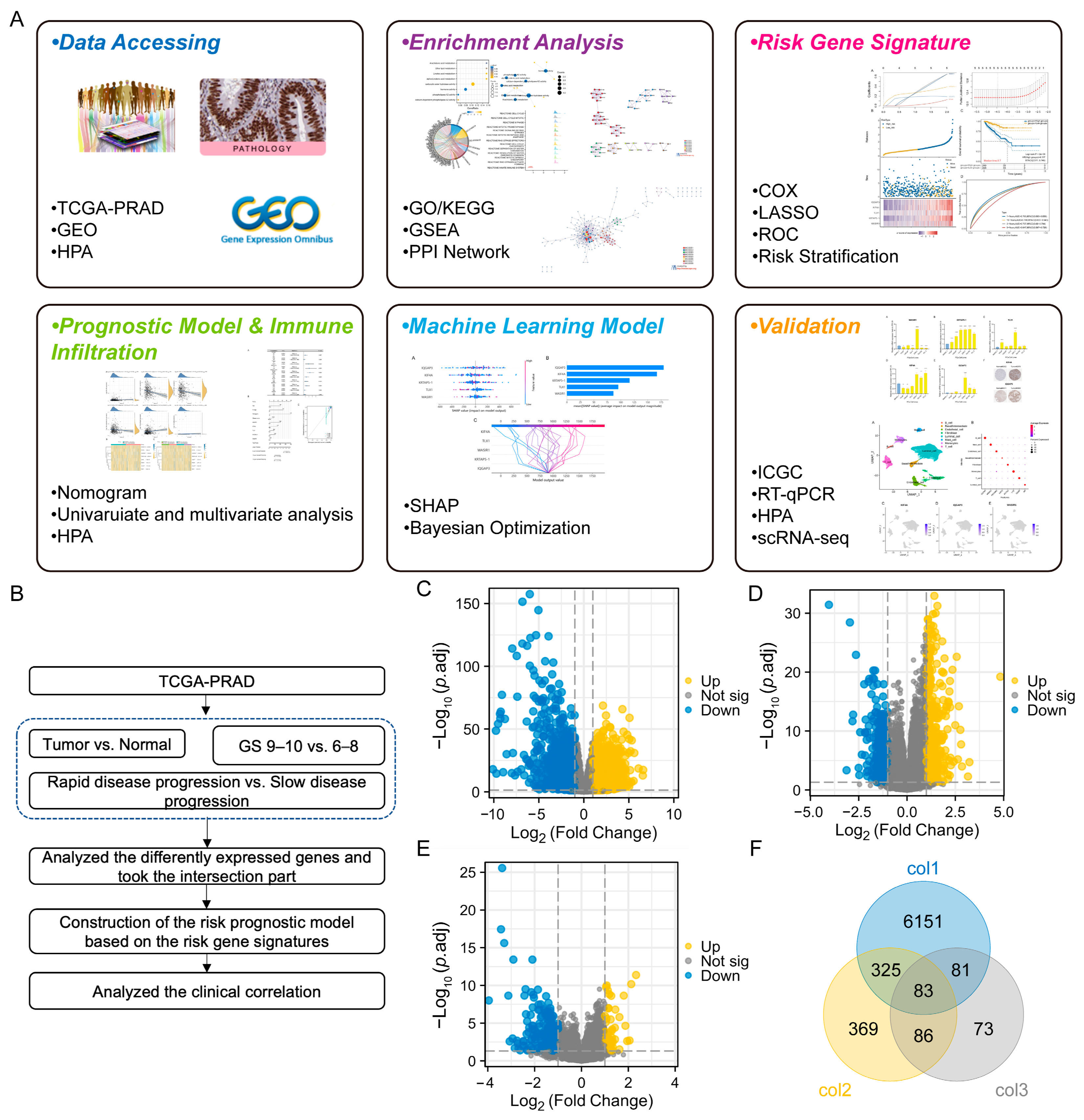{kind=link}
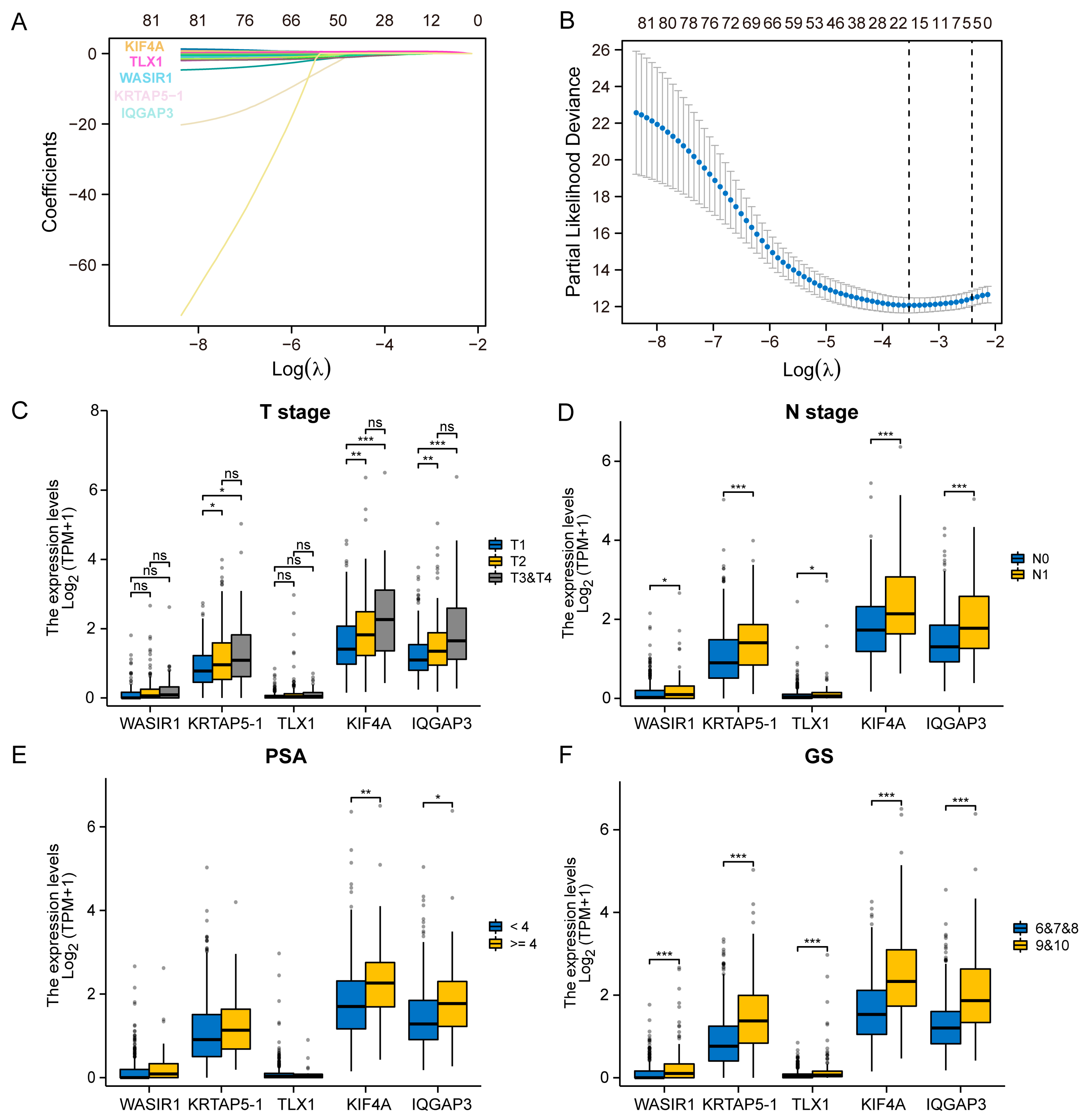{kind=link}
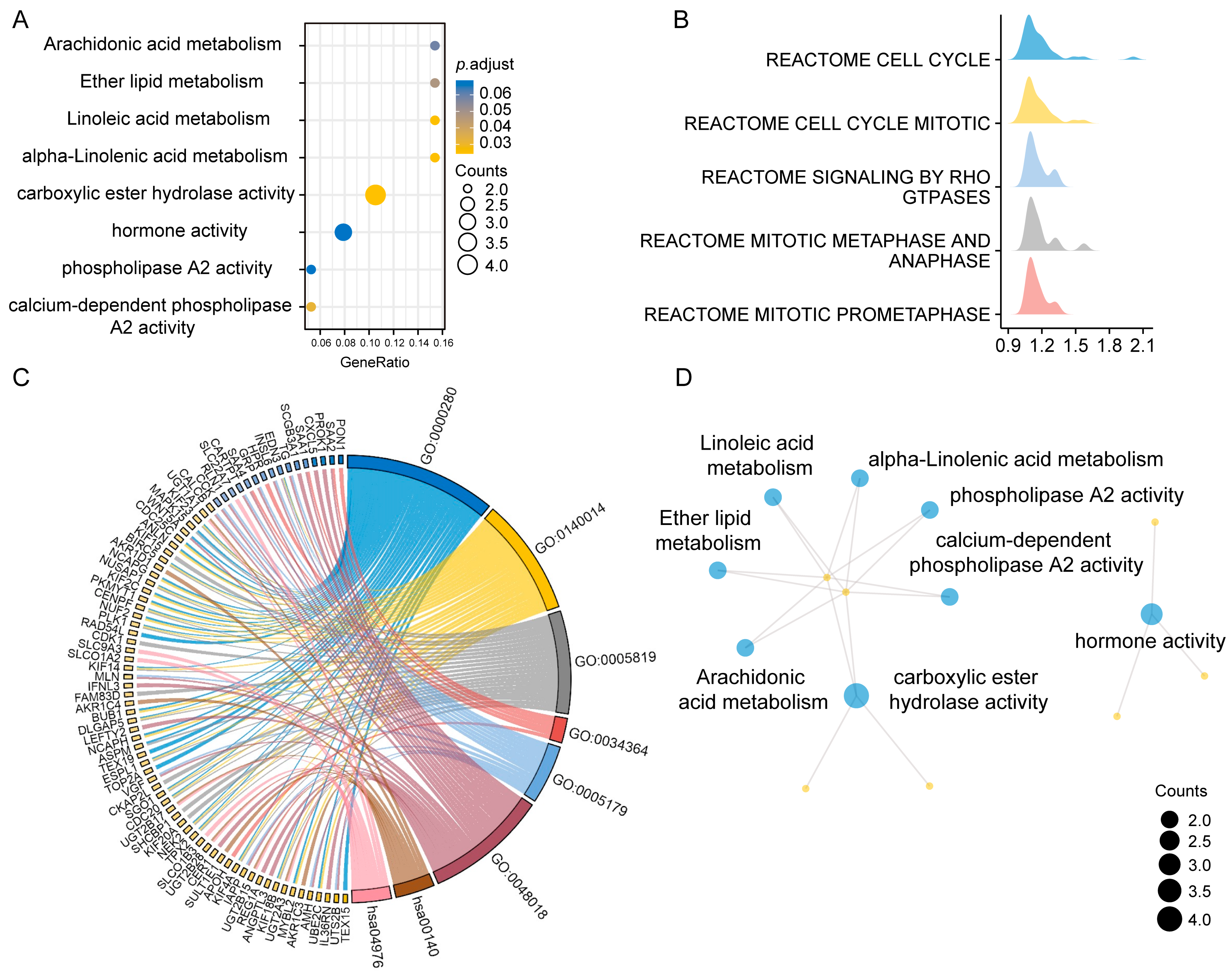{kind=link}
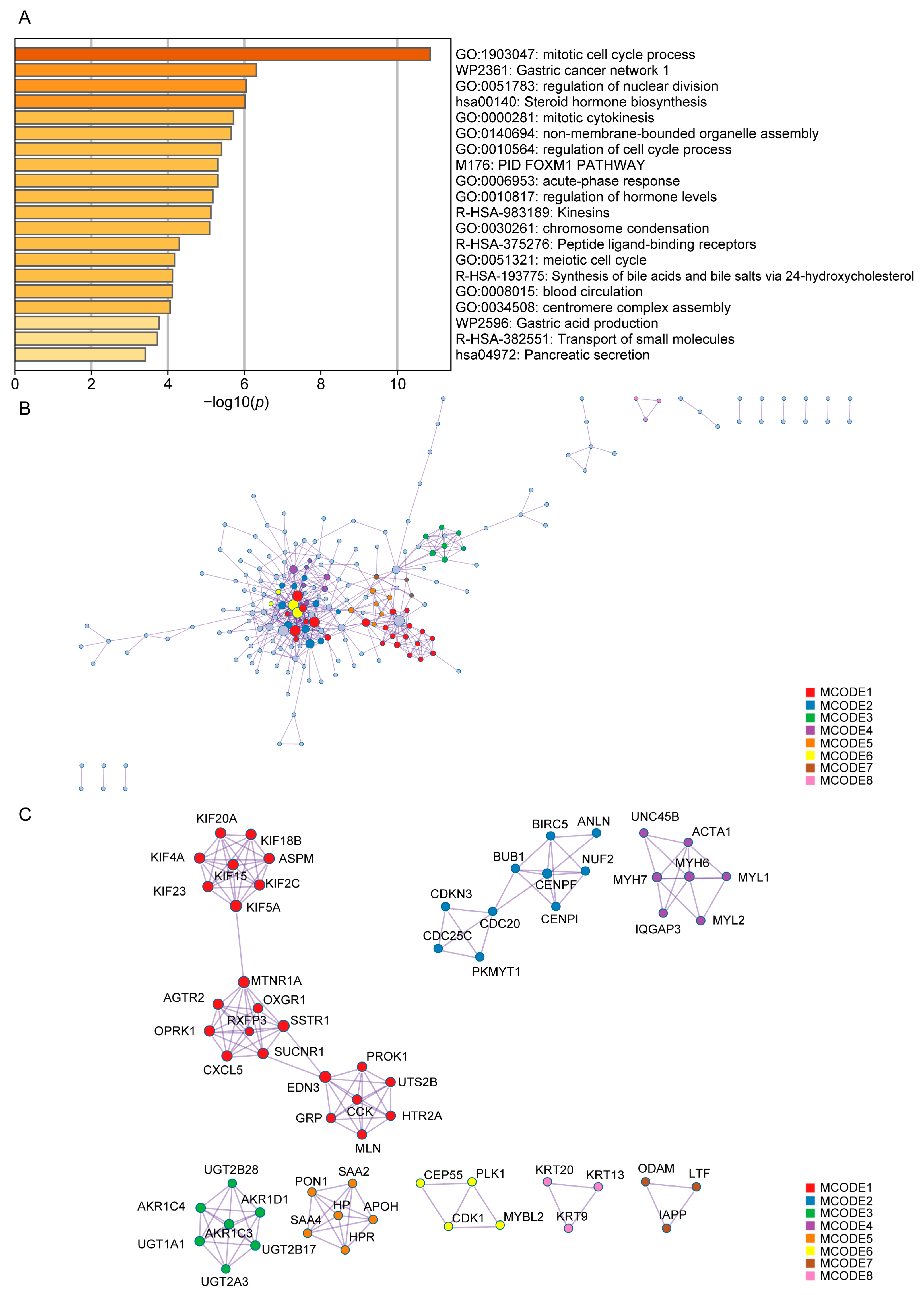{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing Data Access
2.2. Difference Expression of Gene Analysis
2.3. Gene Enrichment Analysis
2.4. Construction of the Protein–Protein Interaction (PPI) Network
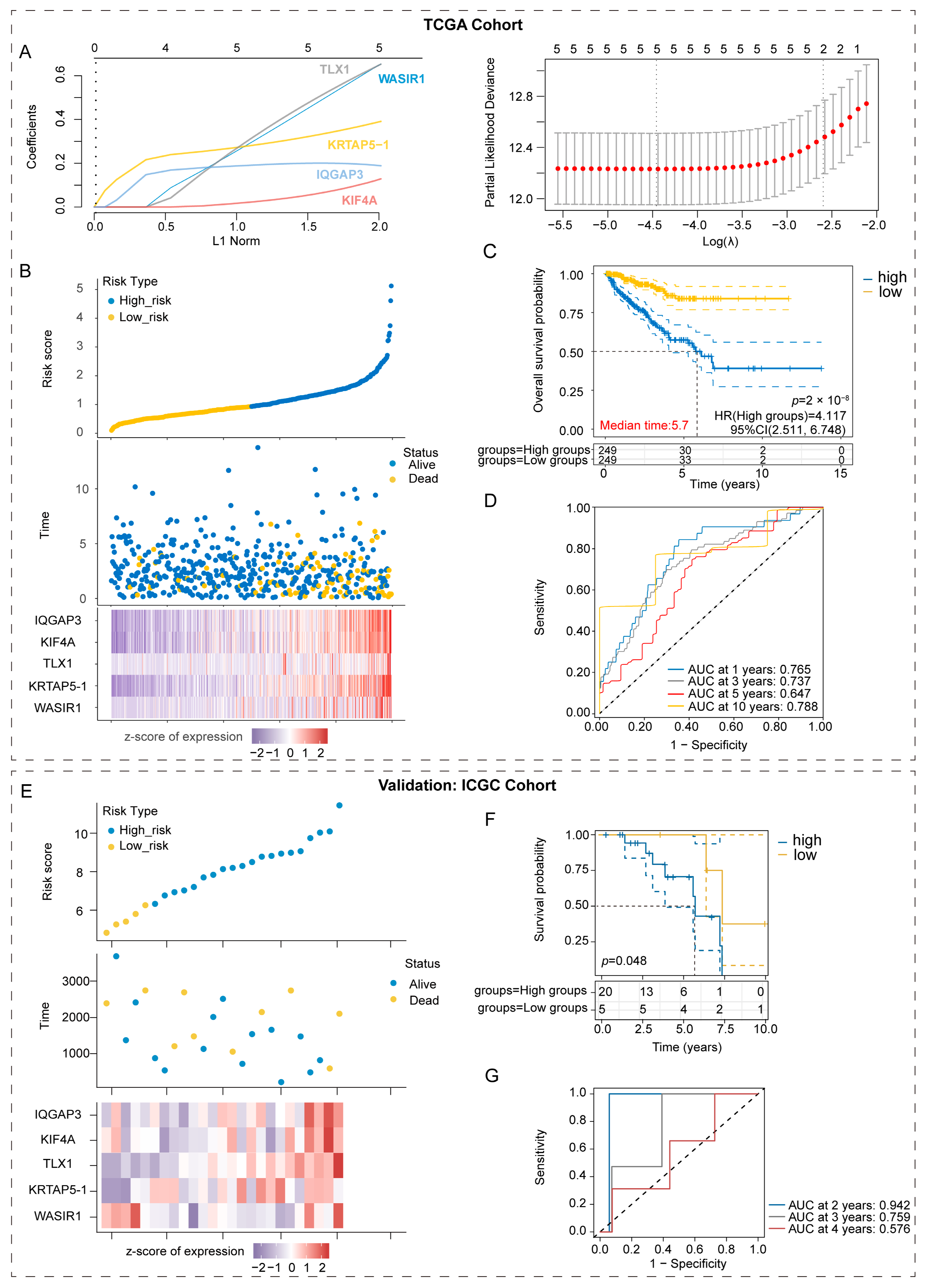
2.5. Construction and Validation of the Prognostic Model Based on the Risk Gene Signatures
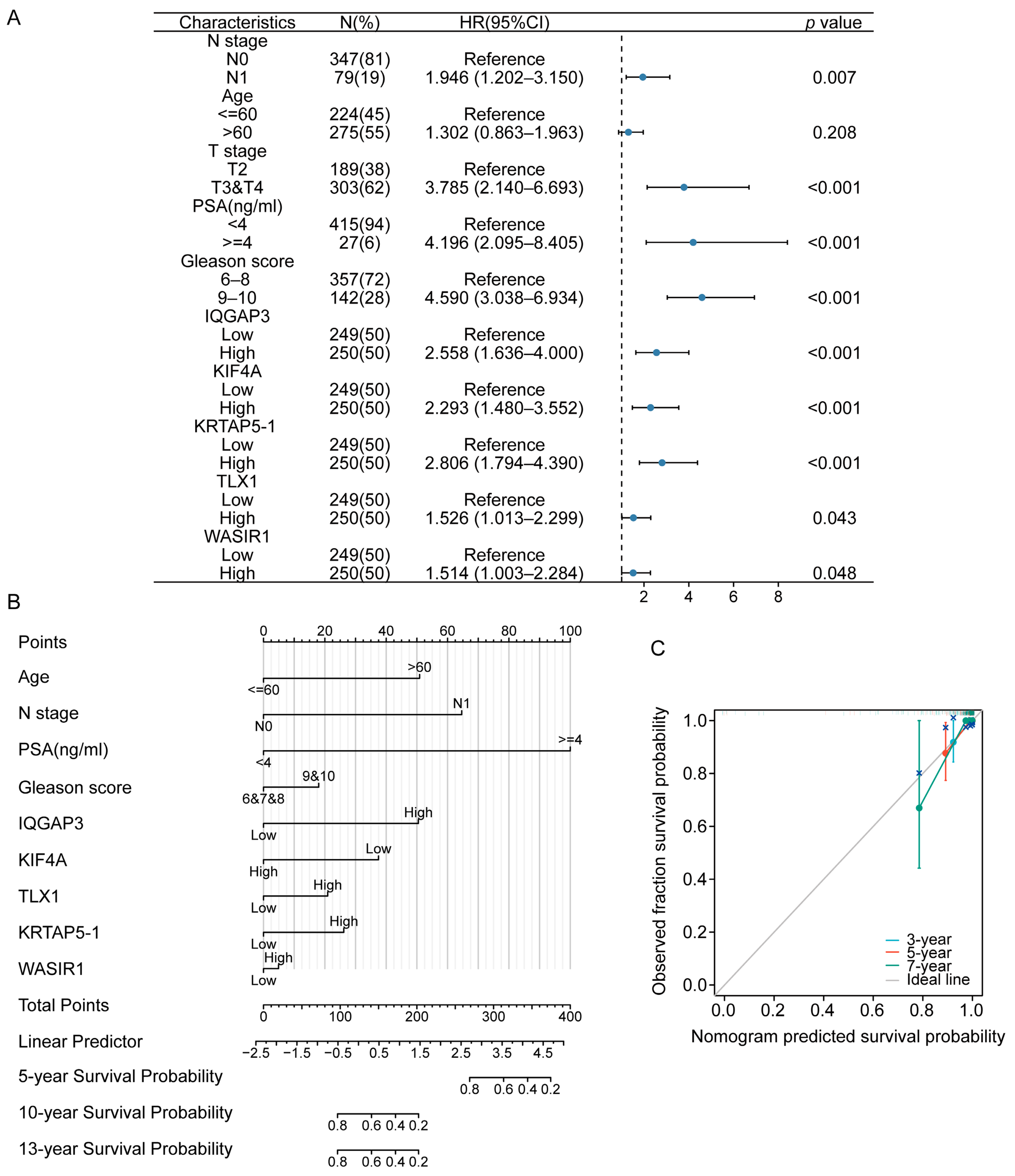
2.6. Construction of the Univariable COX Regression Model and Nomogram
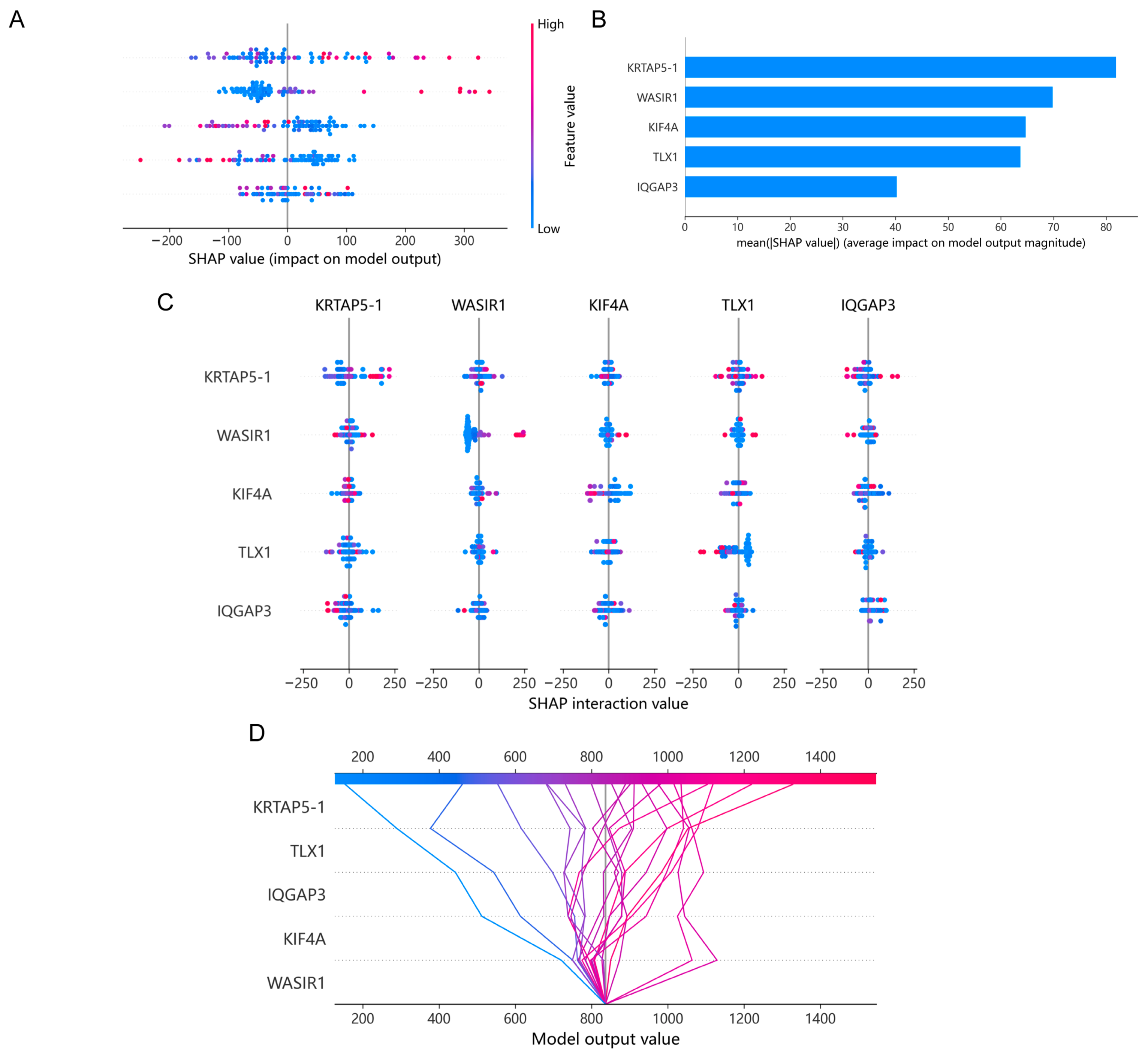
2.7. Machine Learning Model
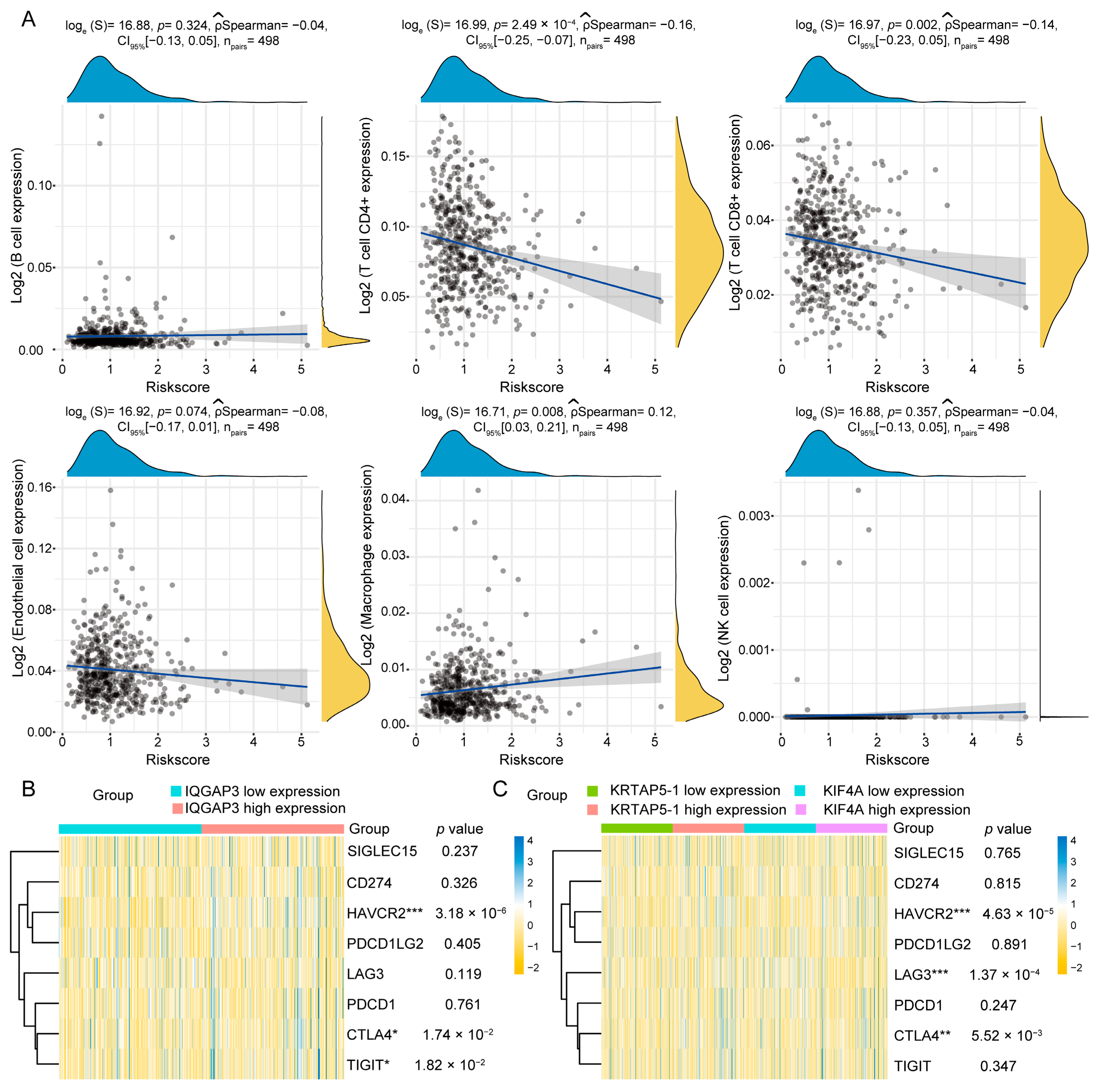
2.8. Correlation Analysis of Immune Infiltration and Genes Associated with ICIs
2.9. Verification of Risk Gene Signature Expression Levels
2.10. Single-Cell RNA Sequencing Data Analysis
2.11. Statistical Analysis
3. Results
3.1. Analysis and Screening of Differentially Expressed Genes (DEGs) of PCa Patients in Three Cohorts
3.2. Selection of the Prognostic Genes and Analysis of the Clinical Correlation
3.3. Enrichment Analysis of the 83 Genes from Three Cohorts and Prediction of Malignant Biological Behavior in PCa Patients with GS 9–10
3.4. Construction of the PPI Network for PCa Patients with GS 9–10
3.5. Construction and Validation of the Prognostic Models Based on Risk Gene Signatures
3.6. Using a Machine Learning Model to Analyze the Impact of Risk Molecules’ Features on Disease Progression in PCa Patients
3.7. Correlation Analysis of Risk Prognostic Model and Immune Treatment Response
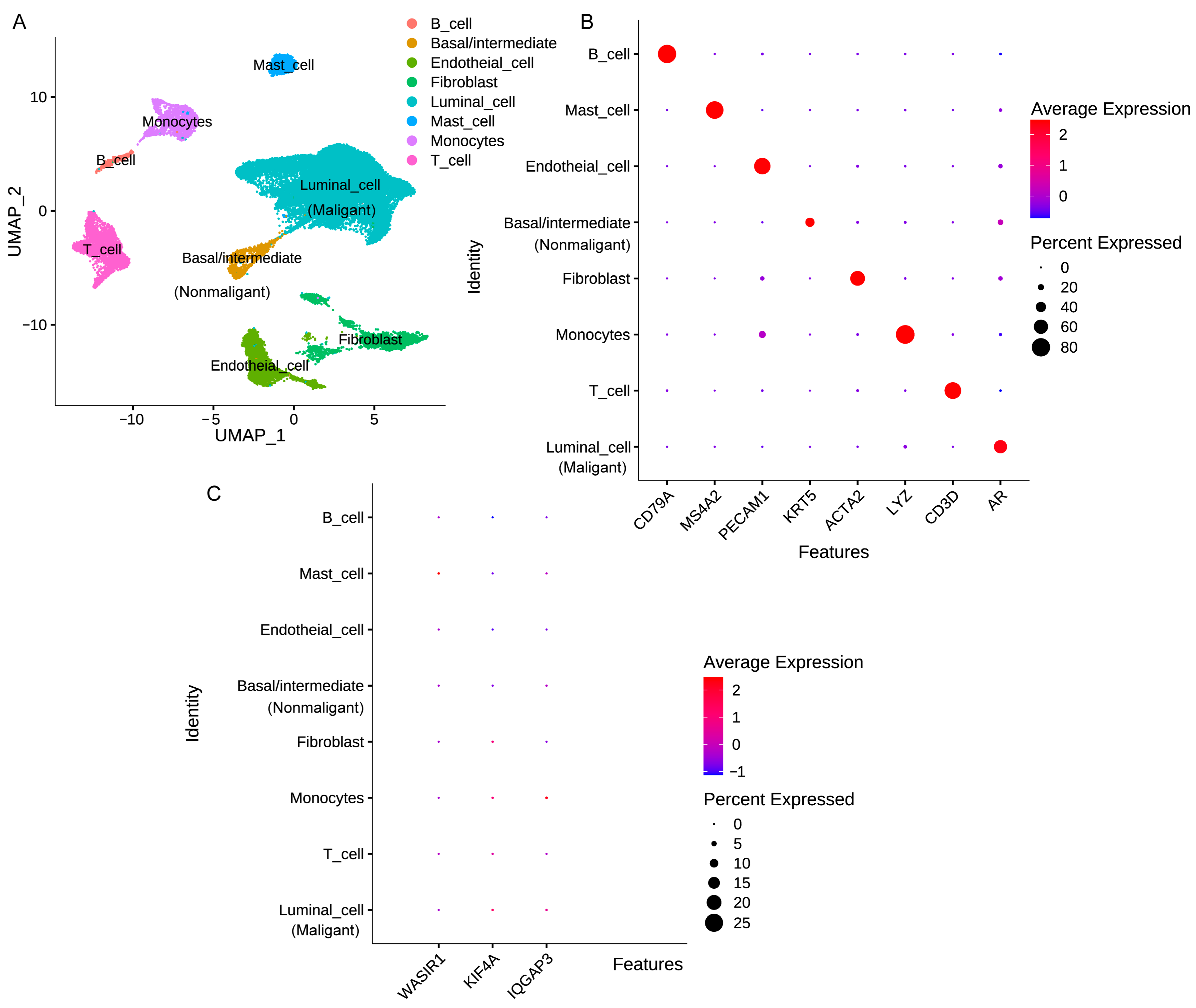
3.8. Analysis of Risk Factor Expression in Different Cellular Subpopulations of PCa Tissue
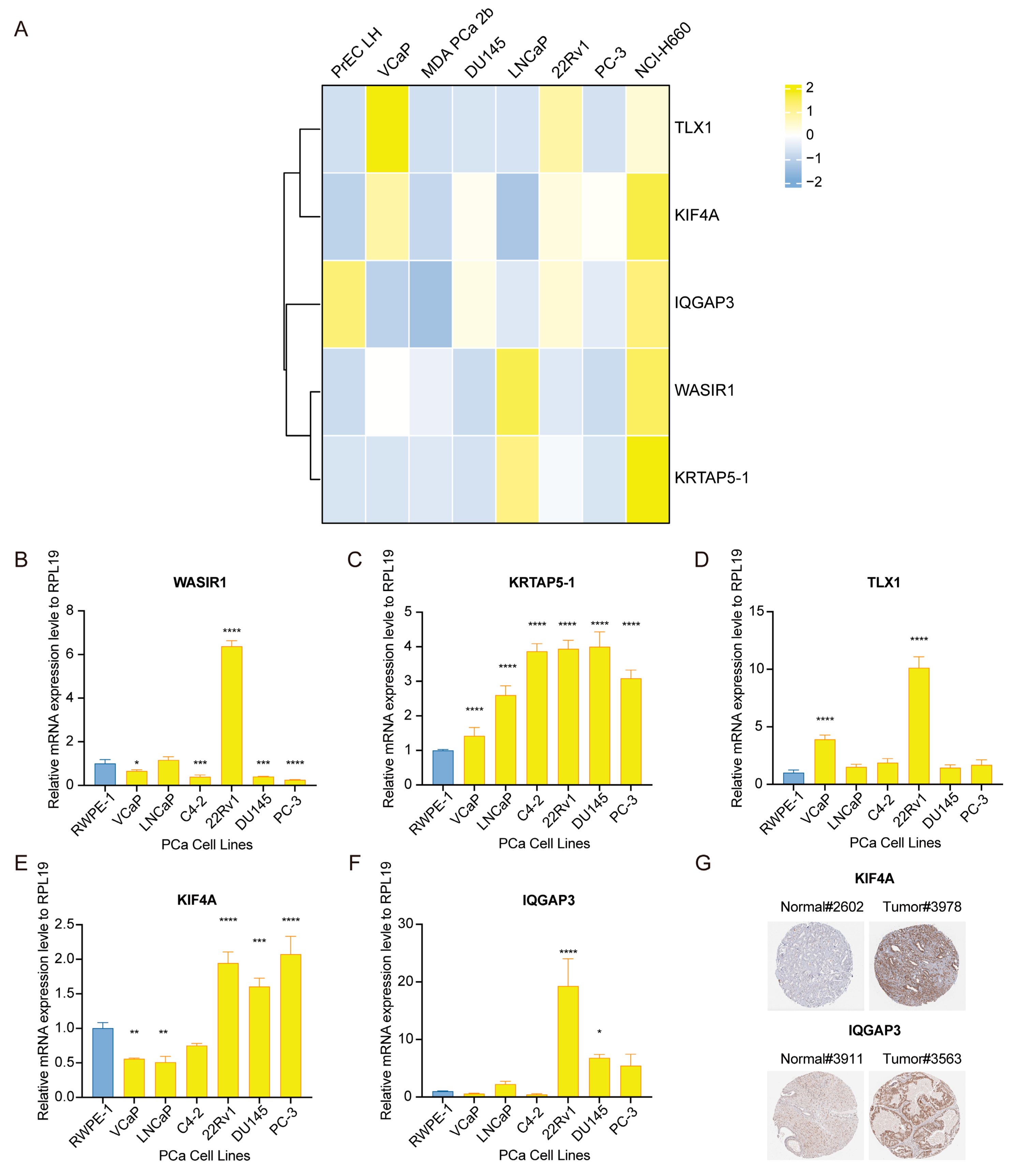
3.9. Assessment of Risk-Associated Gene Signature Expression Patterns in PCa Specimens and Cultures
4. Discussion
5. Conclusions and Future Work
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen Deprivation Therapy |
| AUC | Area Under Curve |
| CRPC | Castration-Resistant Prostate Cancer |
| DEGs | Differentially Expressed Genes |
| EPIC | Estimating the Proportion of Immune and Cancer Cell |
| GS | Gleason Score |
| FDR | False Discovery Rate |
| GSEA | Gene Set Enrichment Analysis |
| HPA | The Human Protein Atlas |
| ICGC | International Cancer Genome Consortium |
| HR | Hazard Ratio |
| ICIs | Immune Checkpoint Inhibitors |
| KM | Kaplan-Meier |
| LASSO | Least Absolute Selection and Shrinkage Operator |
| MCODE | Molecular Complex Detection |
| ORR | Objective Response Rate |
| OS | Overall Survival |
| PCa | Prostate Cancer |
| PD-L1 | Programmed Death Ligand 1 |
| PFS | Progression-Free Survival |
| PRAD | Prostate Adenocarcinoma Database |
| PSA | Prostate-Specific Antigen |
| ROC | Receiver Operating Characteristic |
| RP | Radical Prostatectomy |
| RT-qPCR | Real-Time quantitative Polymerase Chain Reaction |
| scRNA-seq | Single-cell RNA-sequencing |
| TCGA | The Cancer Genome Atlas |
| TMB | Tumor Mutational Burden |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mazariego, C.G.; Egger, S.; King, M.T.; Juraskova, I.; Woo, H.; Berry, M.; Armstrong, B.K.; Smith, D.P. Fifteen year quality of life outcomes in men with localised prostate cancer: Population based Australian prospective study. BMJ 2020, 371, m3503. [Google Scholar] [CrossRef] [PubMed]
- Kishan, A.U.; Cook, R.R.; Ciezki, J.P.; Ross, A.E.; Pomerantz, M.M.; Nguyen, P.L.; Shaikh, T.; Tran, P.T.; Sandler, K.A.; Stock, R.G.; et al. Radical Prostatectomy, External Beam Radiotherapy, or External Beam Radiotherapy with Brachytherapy Boost and Disease Progression and Mortality in Patients with Gleason Score 9-10 Prostate Cancer. JAMA 2018, 319, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Mahal, B.A.; Muralidhar, V.; Martin, N.E.; Orio, P.F.; Mouw, K.W.; King, M.T.; Choueiri, T.K.; Trinh, Q.D.; Hoffman, K.E.; et al. Androgen Deprivation Therapy and Overall Survival for Gleason 8 versus Gleason 9-10 Prostate Cancer. Eur. Urol. 2019, 75, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Deek, M.P.; Yu, C.; Phillips, R.; Song, D.Y.; Deville, C.; Greco, S.; DeWeese, T.L.; Antonarakis, E.S.; Markowski, M.; Paller, C.; et al. Radiation Therapy in the Definitive Management of Oligometastatic Prostate Cancer: The Johns Hopkins Experience. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Nader, R.; El Amm, J.; Aragon-Ching, J.B. Role of chemotherapy in prostate cancer. Asian J. Androl. 2018, 20, 221–229. [Google Scholar] [CrossRef]
- El-Amm, J.; Aragon-Ching, J.B. The Current Landscape of Treatment in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2019, 13, 1179554919833927. [Google Scholar] [CrossRef]
- Jayaraj, R.; Raymond, G.; Krishnan, S.; Tzou, K.S.; Baxi, S.; Ram, M.R.; Govind, S.K.; Chandramoorthy, H.C.; Abu-Khzam, F.N.; Shaw, P. Clinical Theragnostic Potential of Diverse miRNA Expressions in Prostate Cancer: A Systematic Review and Meta-Analysis. Cancers 2020, 12, 1199. [Google Scholar] [CrossRef]
- Jaafar, L.; Fakhoury, I.; Saab, S.; El-Hajjar, L.; Abou-Kheir, W.; El-Sibai, M. StarD13 differentially regulates migration and invasion in prostate cancer cells. Hum. Cell 2021, 34, 607–623. [Google Scholar] [CrossRef]
- Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers 2019, 11, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucchiara, V.; Cooperberg, M.R.; Dall’Era, M.; Lin, D.W.; Montorsi, F.; Schalken, J.A.; Evans, C.P. Genomic Markers in Prostate Cancer Decision Making. Eur. Urol. 2018, 73, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.Y.; Huang, H.C.; Spratt, D.E.; Zhao, S.G.; Sandler, H.M.; Simko, J.P.; Davicioni, E.; Nguyen, P.L.; Pollack, A.; Efstathiou, J.A.; et al. Validation of a 22-Gene Genomic Classifier in Patients with Recurrent Prostate Cancer: An Ancillary Study of the NRG/RTOG 9601 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 544–552. [Google Scholar] [CrossRef]
- Dalela, D.; Löppenberg, B.; Sood, A.; Sammon, J.; Abdollah, F. Contemporary Role of the Decipher® Test in Prostate Cancer Management: Current Practice and Future Perspectives. Rev. Urol. 2016, 18, 1–9. [Google Scholar]
- Grönberg, H.; Adolfsson, J.; Aly, M.; Nordström, T.; Wiklund, P.; Brandberg, Y.; Thompson, J.; Wiklund, F.; Lindberg, J.; Clements, M.; et al. Prostate cancer screening in men aged 50-69 years (STHLM3): A prospective population-based diagnostic study. Lancet Oncol. 2015, 16, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Qin, S.; Luo, S.; Li, Z.; Cheng, Y.; Fan, Y.; Sun, Y.; Yin, X.; Yuan, X.; Li, W.; et al. Pembrolizumab versus chemotherapy for patients with esophageal squamous cell carcinoma enrolled in the randomized KEYNOTE-181 trial in Asia. ESMO Open 2022, 7, 100341. [Google Scholar] [CrossRef]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: Results of >2 years of follow-up. Ann. Oncol. 2019, 30, 970–976. [Google Scholar] [CrossRef]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.H.; Doi, T.; Moriwaki, T.; Kim, S.B.; Lee, S.H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Spigel, D.R.; Faivre-Finn, C.; Gray, J.E.; Vicente, D.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Garassino, M.C.; Hui, R.; Quantin, X.; et al. Five-Year Survival Outcomes from the PACIFIC Trial: Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 1301–1311. [Google Scholar] [CrossRef]
- Yu, E.Y.; Kolinsky, M.P.; Berry, W.R.; Retz, M.; Mourey, L.; Piulats, J.M.; Appleman, L.J.; Romano, E.; Gravis, G.; Gurney, H.; et al. Pembrolizumab Plus Docetaxel and Prednisone in Patients with Metastatic Castration-resistant Prostate Cancer: Long-term Results from the Phase 1b/2 KEYNOTE-365 Cohort B Study. Eur. Urol. 2022, 82, 22–30. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Knipper, S.; Palumbo, C.; Pecoraro, A.; Rosiello, G.; Tian, Z.; Briganti, A.; Zorn, K.C.; Saad, F.; Tilki, D.; Graefen, M.; et al. Survival outcomes of radical prostatectomy vs. external beam radiation therapy in prostate cancer patients with Gleason Score 9-10 at biopsy: A population-based analysis. Urol. Oncol. 2020, 38, 79.e9–79.e14. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Cheng, X.; Zeng, Z.; Yang, H.; Chen, Y.; Liu, Y.; Zhou, X.; Zhang, C.; Wang, G. Novel cuproptosis-related long non-coding RNA signature to predict prognosis in prostate carcinoma. BMC Cancer 2023, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.L.; Liu, C.H.; Wang, S.C.; Huang, S.P.; Cho, Y.C.; Bao, B.Y.; Su, C.C.; Yeh, H.C.; Lee, C.H.; Teng, P.C.; et al. Identification of a Steroid Hormone-Associated Gene Signature Predicting the Prognosis of Prostate Cancer through an Integrative Bioinformatics Analysis. Cancers 2022, 14, 1565. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, X.; Ma, C.; Hu, J. m(6)A Methylation Regulators Are Predictive Biomarkers for Tumour Metastasis in Prostate Cancer. Cancers 2022, 14, 4035. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; Yan, Y.; Wang, C.; Colic, M.; Olszewski, K.; Horbath, A.; Chen, X.; Lei, G.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Tang, H.; Mi, Y.; Zhu, Y.; Wan, F.; Ye, D. A novel gene signature to predict immune infiltration and outcome in patients with prostate cancer. Oncoimmunology 2020, 9, 1762473. [Google Scholar] [CrossRef]
- Alhasan, A.H.; Scott, A.W.; Wu, J.J.; Feng, G.; Meeks, J.J.; Thaxton, C.S.; Mirkin, C.A. Circulating microRNA signature for the diagnosis of very high-risk prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 10655–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Cazorla-Vázquez, S.; Ferrazzi, F.; Wiederstein, J.L.; Gründl, M.; Weinstock, G.; Vergarajauregui, S.; Eckstein, M.; Krüger, M.; Gaubatz, S.; et al. IQGAP3, a YAP Target, Is Required for Proper Cell-Cycle Progression and Genome Stability. Mol. Cancer Res. 2021, 19, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef] [Green Version]
- Oue, N.; Sentani, K.; Sakamoto, N.; Uraoka, N.; Yasui, W. Molecular carcinogenesis of gastric cancer: Lauren classification, mucin phenotype expression, and cancer stem cells. Int. J. Clin. Oncol. 2019, 24, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The biology of IQGAP proteins: Beyond the cytoskeleton. EMBO Rep. 2015, 16, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Watanabe, T.; Noritake, J.; Fukata, M.; Yoshimura, T.; Itoh, N.; Harada, T.; Nakagawa, M.; Matsuura, Y.; Arimura, N.; et al. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. J. Cell Sci. 2007, 120, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, J.; Douchi, D.; Myint, K.; Mon, N.N.; Yamamura, A.; Kohu, K.; Heng, D.L.; Chen, S.; Mawan, N.A.; Nuttonmanit, N.; et al. Iqgap3-Ras axis drives stem cell proliferation in the stomach corpus during homoeostasis and repair. Gut 2021, 70, 1833–1846. [Google Scholar] [CrossRef]
- Dongol, S.; Zhang, Q.; Qiu, C.; Sun, C.; Zhang, Z.; Wu, H.; Kong, B. IQGAP3 promotes cancer proliferation and metastasis in high-grade serous ovarian cancer. Oncol. Lett. 2020, 20, 1179–1192. [Google Scholar] [CrossRef]
- Hua, X.; Long, Z.Q.; Guo, L.; Wen, W.; Huang, X.; Zhang, W.W. IQGAP3 Overexpression Correlates with Poor Prognosis and Radiation Therapy Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 584450. [Google Scholar] [CrossRef]
- Handy, C.E.; Antonarakis, E.S. Sipuleucel-T for the treatment of prostate cancer: Novel insights and future directions. Future Oncol. 2018, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients with Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, F.; Gao, X.; Ma, M.; Xie, M.; Shang, S.; Ren, X.; Liu, M.; Chen, J. Crafting a Personalized Prognostic Model for Malignant Prostate Cancer Patients Using Risk Gene Signatures Discovered through TCGA-PRAD Mining, Machine Learning, and Single-Cell RNA-Sequencing. Diagnostics 2023, 13, 1997. https://doi.org/10.3390/diagnostics13121997
Lyu F, Gao X, Ma M, Xie M, Shang S, Ren X, Liu M, Chen J. Crafting a Personalized Prognostic Model for Malignant Prostate Cancer Patients Using Risk Gene Signatures Discovered through TCGA-PRAD Mining, Machine Learning, and Single-Cell RNA-Sequencing. Diagnostics. 2023; 13(12):1997. https://doi.org/10.3390/diagnostics13121997
Chicago/Turabian StyleLyu, Feng, Xianshu Gao, Mingwei Ma, Mu Xie, Shiyu Shang, Xueying Ren, Mingzhu Liu, and Jiayan Chen. 2023. "Crafting a Personalized Prognostic Model for Malignant Prostate Cancer Patients Using Risk Gene Signatures Discovered through TCGA-PRAD Mining, Machine Learning, and Single-Cell RNA-Sequencing" Diagnostics 13, no. 12: 1997. https://doi.org/10.3390/diagnostics13121997
APA StyleLyu, F., Gao, X., Ma, M., Xie, M., Shang, S., Ren, X., Liu, M., & Chen, J. (2023). Crafting a Personalized Prognostic Model for Malignant Prostate Cancer Patients Using Risk Gene Signatures Discovered through TCGA-PRAD Mining, Machine Learning, and Single-Cell RNA-Sequencing. Diagnostics, 13(12), 1997. https://doi.org/10.3390/diagnostics13121997