


A First Computational Frame for Recognizing Heparin-Binding Protein



Abstract
:1. Introduction
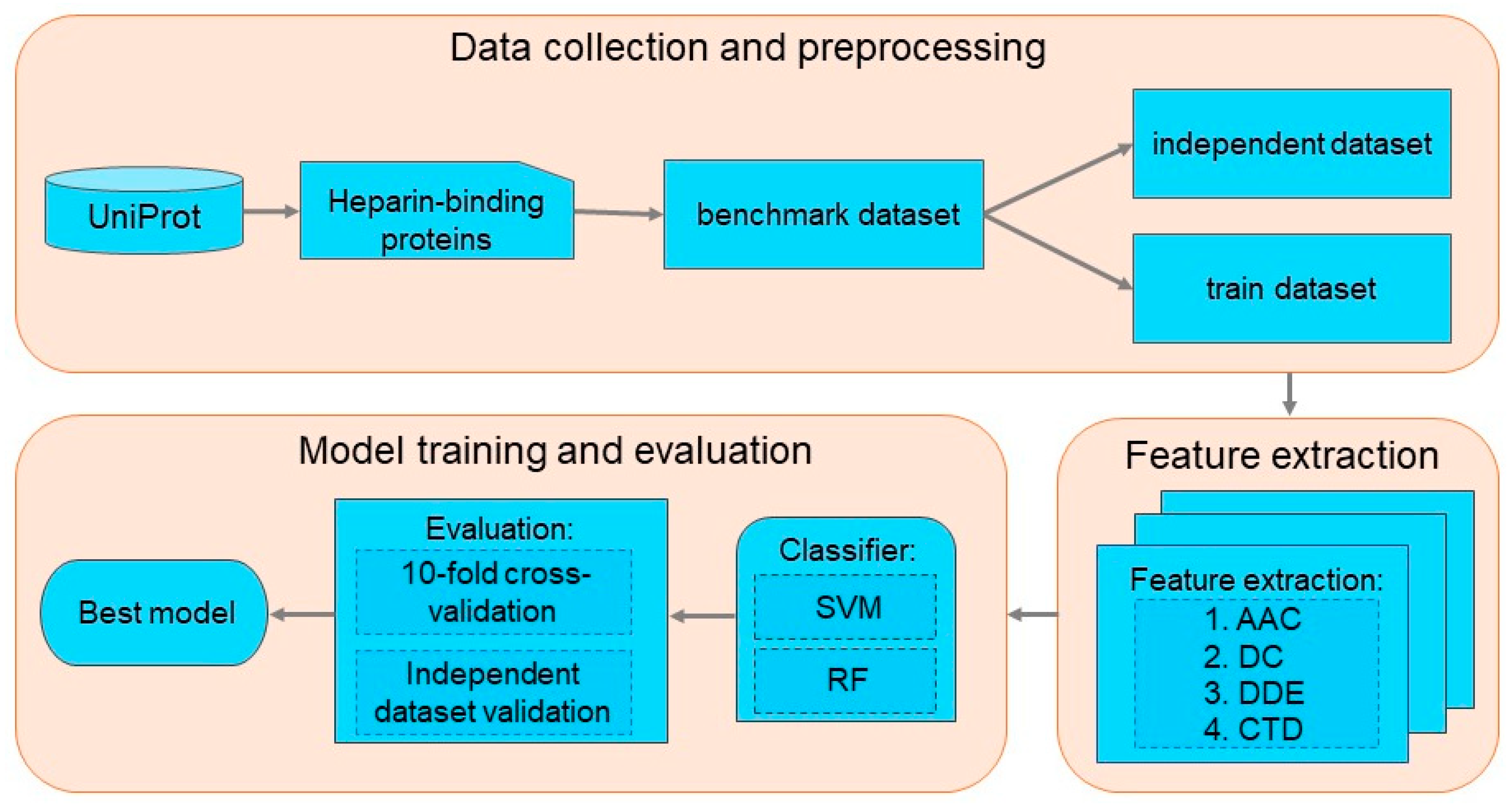
2. Materials and Methods
2.1. Benchmark Dataset Construction
2.2. Formulation of Protein Sequences
2.2.1. Amino Acid Composition (AAC)
2.2.2. Dipeptide Composition (DC)
2.2.3. Dipeptide Deviation from Expected Mean (DDE)
2.2.4. Composition/Transition/Distribution (CTD)
2.3. Machine Learning Methods
2.4. Evaluation Indexes
3. Results
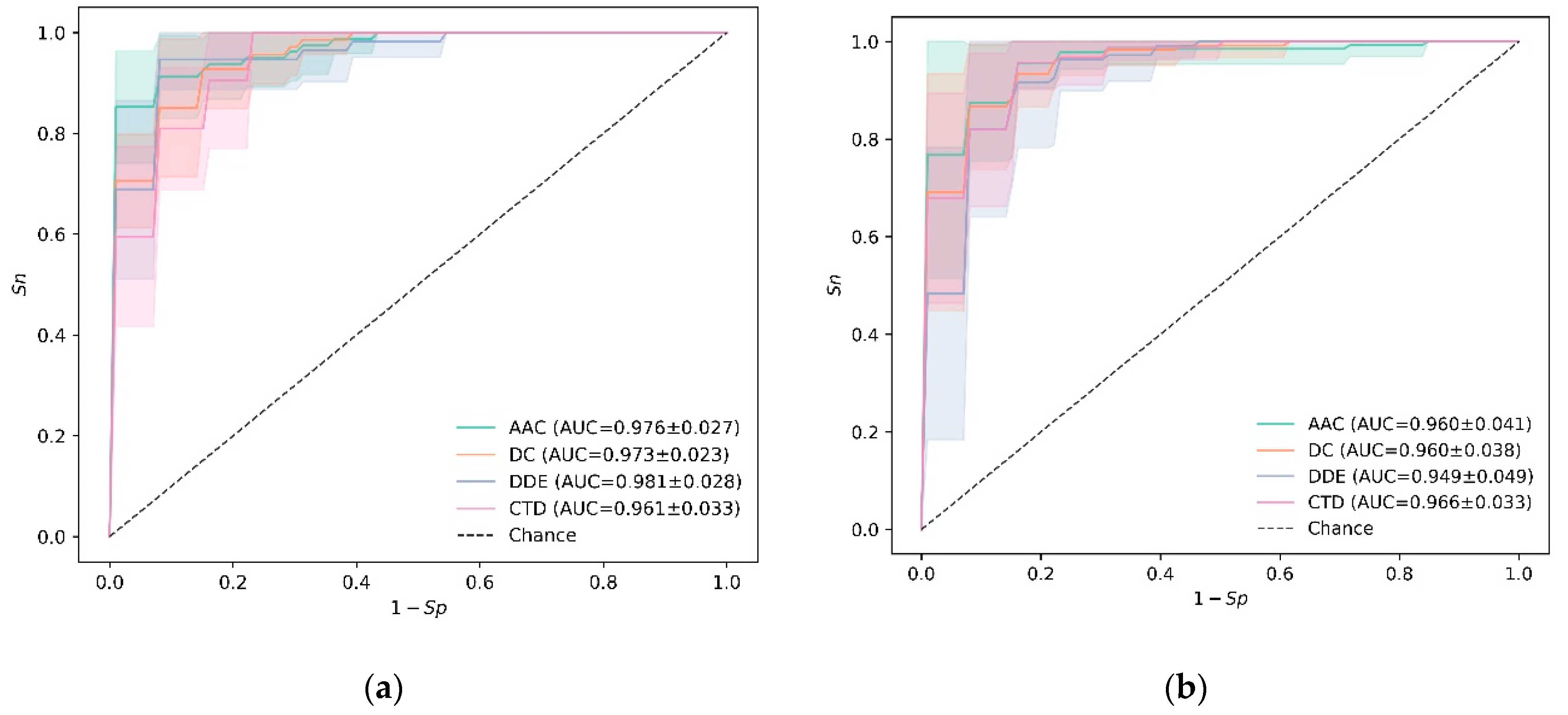
3.1. Experiments on Training Data
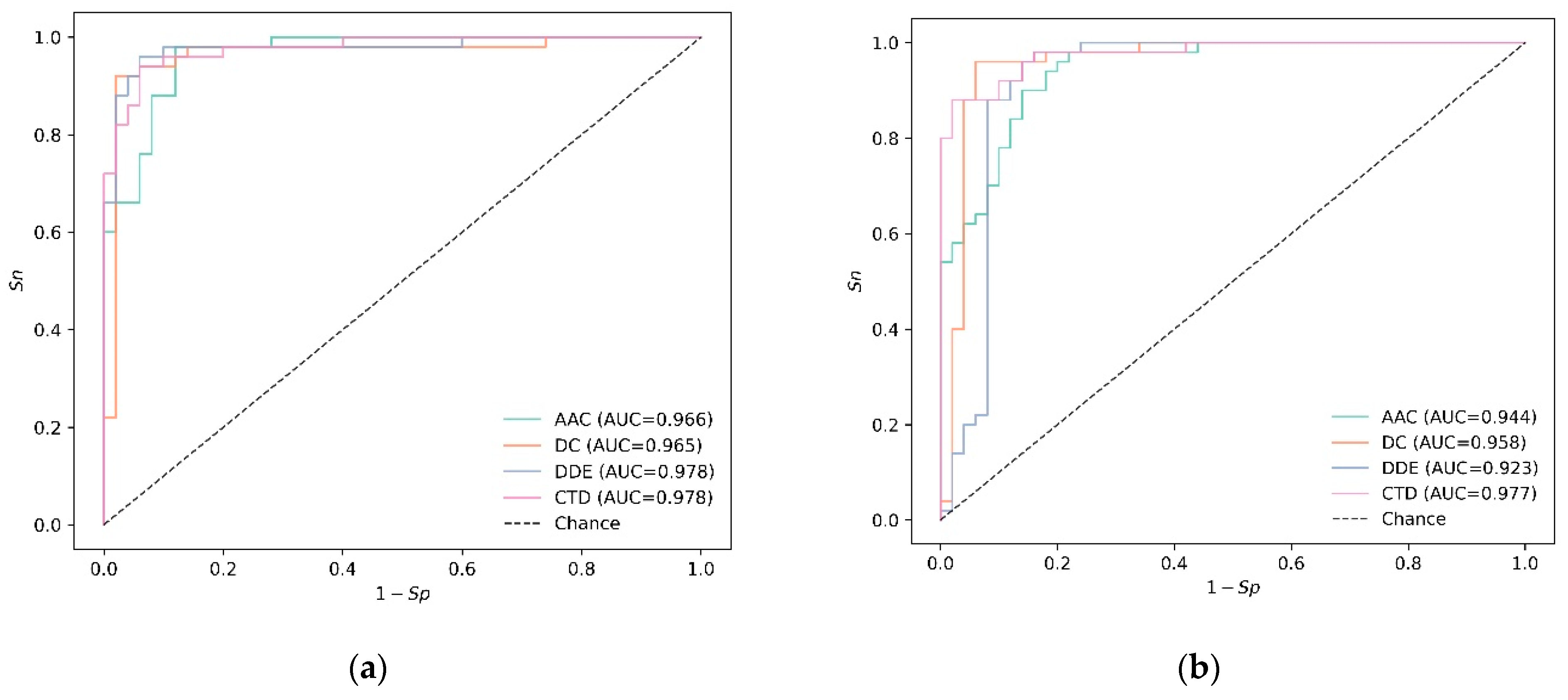
3.2. Experiments on Independent Data
4. Further Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| auROC | area under the receiver operating characteristic curve |
| AAC | amino acid composition |
| CTD | composition/transition/distribution |
| DC | dipeptide composition |
| DDE | dipeptide deviation from expected mean |
| HBP | heparin-binding protein |
| MCC | Matthews correlation coefficient |
| OA | overall accuracy |
| PseAAC | pseudo amino acid composition |
| RAAC | reduced amino acid composition |
| RF | random forest |
| Sn | sensitivity |
| Sp | specificity |
| SVM | support vector machine |
| UniProt | Universal Protein Resource |
References
- Fisher, J.; Kahn, F.; Wiebe, E.; Gustafsson, P.; Kander, T.; Mellhammar, L.; Bentzer, P.; Linder, A. The Dynamics of Circulating Heparin-Binding Protein: Implications for Its Use as a Biomarker. J. Innate. Immun. 2022, 14, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Qi, C.; Zhuang, H.; Fu, T.; Zhang, X. gutMDisorder: A comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 2020, 48, D554–D560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Shen, Z.-A.; Zhou, Y.-K.; Du, P.-F. Recent advances in predicting protein-lncRNA interactions using machine learning methods. Curr. Gene Ther. 2021, 22, 228–244. [Google Scholar]
- Yang, Y.; Liu, G.; He, Q.; Shen, J.; Xu, L.; Zhu, P.; Zhao, M. A Promising Candidate: Heparin-Binding Protein Steps onto the Stage of Sepsis Prediction. J. Immunol. Res. 2019, 2019, 7515346. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Qi, C.; Yang, H.; Lu, M.; Cai, Y.; Fu, T.; Ren, J.; Jin, Q.; Zhang, X. gutMGene: A comprehensive database for target genes of gut microbes and microbial metabolites. Nucleic Acids Res. 2021, 50, D795–D800. [Google Scholar] [CrossRef]
- Sato, W.; Kadomatsu, K.; Yuzawa, Y.; Muramatsu, H.; Hotta, N.; Matsuo, S.; Muramatsu, T. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J. Immunol. 2001, 167, 3463–3469. [Google Scholar] [CrossRef]
- Ao, C.; Yu, L.; Zou, Q. Prediction of bio-sequence modifications and the associations with diseases. Brief. Funct. Genom. 2021, 20, 1–18. [Google Scholar] [CrossRef]
- Qi, C.; Wang, C.; Zhao, L.; Zhu, Z.; Wang, P.; Zhang, S.; Cheng, L.; Zhang, X. SCovid: Single-cell atlases for exposing molecular characteristics of COVID-19 across 10 human tissues. Nucleic Acids Res. 2021, 50, D867–D874. [Google Scholar] [CrossRef]
- Bascuas, T.; Zedira, H.; Kropp, M.; Harmening, N.; Asrih, M.; Prat-Souteyrand, C.; Tian, S.; Thumann, G. Human Retinal Pigment Epithelial Cells Overexpressing the Neuroprotective Proteins PEDF and GM-CSF to Treat Degeneration of the Neural Retina. Curr. Gene Ther. 2021, 22, 168–183. [Google Scholar] [CrossRef]
- Ning, L.; Abagna, H.B.; Jiang, Q.; Liu, S.; Huang, J. Development and application of therapeutic antibodies against COVID-19. Int. J. Biol. Sci. 2021, 17, 1486–1496. [Google Scholar] [CrossRef]
- Neumann, A. Rapid release of sepsis markers heparin-binding protein and calprotectin triggered by anaerobic cocci poses an underestimated threat. Anaerobe 2022, 75, 102584. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Liu, M.; Gou, Y.; Yang, Y.; He, B.; Huang, J. Development and application of ribonucleic acid therapy strategies against COVID-19. Int. J. Biol. Sci. 2022, 18, 5070–5085. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Xu, Y.; Ning, L.; Pan, X.; Li, Y.; Zhao, Q.; Pang, B.; Huang, J.; Deng, K.; Zhang, Y. TCM2COVID: A resource of anti-COVID-19 traditional Chinese medicine with effects and mechanisms. iMETA 2022, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Linder, A. Heparin-binding protein: A key player in the pathophysiology of organ dysfunction in sepsis. J. Intern. Med. 2017, 281, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Yo, C.H.; Hsu, W.T.; Qian, F.; Wu, B.S.; Dou, Q.L.; Lee, C.C. Accuracy of Heparin-Binding Protein in Diagnosing Sepsis: A Systematic Review and Meta-Analysis. Crit. Care Med. 2021, 49, e80–e90. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Wang, J.; Zou, B.; Li, L.; Yao, L.; Chen, K.; Ning, L.; Wu, B.; Zhao, X.; et al. Cellinker: A platform of ligand-receptor interactions for intercellular communication analysis. Bioinformatics 2021, 37, 2025–2032. [Google Scholar] [CrossRef]
- Su, W.; Liu, M.L.; Yang, Y.H.; Wang, J.S.; Li, S.H.; Lv, H.; Dao, F.Y.; Yang, H.; Lin, H. PPD: A Manually Curated Database for Experimentally Verified Prokaryotic Promoters. J. Mol. Biol. 2021, 433, 166860. [Google Scholar] [CrossRef]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Cheng, L.; Hu, Y.; Sun, J.; Zhou, M.; Jiang, Q. DincRNA: A comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 2018, 34, 1953–1956. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.; Zulfiqar, H.; Khan, A.A.; Gul, I.; Dao, F.Y.; Zhang, Z.Y.; Yu, X.L.; Tang, L. iThermo: A Sequence-Based Model for Identifying Thermophilic Proteins Using a Multi-Feature Fusion Strategy. Front. Microbiol. 2022, 13, 790063. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, J.Y.; Zhang, Y.; Zhang, H.; Gao, S.; Wang, T.; Han, Z.; Wang, L.; Sun, B.L.; Liu, G. rs1990622 variant associates with Alzheimer’s disease and regulates TMEM106B expression in human brain tissues. BMC Med. 2021, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, H.; Liu, B.; Gao, S.; Wang, T.; Han, Z.; International Genomics of Alzheimer’s, P.; Ji, X.; Liu, G. rs34331204 regulates TSPAN13 expression and contributes to Alzheimer’s disease with sex differences. Brain 2020, 143, e95. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Zhang, Z.Y.; Lin, H. BDselect: A Package for k-mer Selection Based on the Binomial Distribution. Curr. Bioinform. 2022, 17, 238–244. [Google Scholar]
- Sanami, S.; Alizadeh, M.; Nosrati, M.; Dehkordi, K.A.; Azadegan-Dehkordi, F.; Tahmasebian, S.; Nosrati, H.; Arjmand, M.-H.; Ghasemi-Dehnoo, M.; Rafiei, A.; et al. Exploring SARS-CoV-2 structural proteins to design a multi-epitope vaccine using immunoinformatics approach: An in silico study. Comput. Biol. Med. 2021, 133, 104390. [Google Scholar] [CrossRef]
- Wu, X.; Yu, L. EPSOL: Sequence-based protein solubility prediction using multidimensional embedding. Bioinformatics 2021, 37, 4314–4320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wan, J.; Wang, G. A survey on computational methods in discovering protein inhibitors of SARS-CoV-2. Brief. Bioinform. 2022, 23, bbab416. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, H.; Li, H.; Wu, Y.; Wang, G. Identifying Plant Pentatricopeptide Repeat Proteins Using a Variable Selection Method. Front. Plant. Sci. 2021, 12, 506681. [Google Scholar] [CrossRef]
- Teng, Z.; Zhang, Z.; Tian, Z.; Li, Y.; Wang, G. ReRF-Pred: Predicting amyloidogenic regions of proteins based on their pseudo amino acid composition and tripeptide composition. BMC Bioinform. 2021, 22, 545. [Google Scholar] [CrossRef]
- Zhai, Y.; Chen, Y.; Teng, Z.; Zhao, Y. Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein-Protein Interactions. Front. Cell. Dev. Biol. 2020, 8, 591487. [Google Scholar] [CrossRef]
- Tao, Z.; Li, Y.; Teng, Z.; Zhao, Y. A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 2020, 8926750. [Google Scholar] [CrossRef]
- Hu, Y.; Qiu, S.; Cheng, L. Integration of Multiple-Omics Data to Analyze the Population-Specific Differences for Coronary Artery Disease. Comput. Math. Methods Med. 2021, 2021, 7036592. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Y.; Zhang, H.; Gao, S.; Wang, L.; Wang, T.; Han, Z.; International Genomics of Alzheimer’s, P.; Liu, G. Mendelian randomization highlights causal association between genetically increased C-reactive protein levels and reduced Alzheimer’s disease risk. Alzheimers Dement. 2022, 18, 2003–2006. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Y.; Zhang, H.; Gao, S.; Wang, L.; Wang, T.; Han, Z.; Sun, B.L.; Liu, G. Cognitive performance protects against Alzheimer’s disease independently of educational attainment and intelligence. Mol. Psychiatry 2022, 27, 4297–4306. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, H.-D.; Zulfiqar, H.; Yuan, S.-S.; Huang, Q.-L.; Zhang, Z.-Y.; Deng, K.-J. iBLP: An XGBoost-Based Predictor for Identifying Bioluminescent Proteins. Comput. Math. Methods Med. 2021, 2021, 6664362. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Y.; Chai, L.; Li, Q.; Liu, J.; Lin, H.; Liu, L. A deep learning model to identify gene expression level using cobinding transcription factor signals. Brief. Bioinform. 2022, 23, bbab501. [Google Scholar] [CrossRef]
- Li, H.; Gong, Y.; Liu, Y.; Lin, H.; Wang, G. Detection of transcription factors binding to methylated DNA by deep recurrent neural network. Brief. Bioinform. 2022, 23, bbab533. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Guan, H.; Fan, G.-L. The Regulation of Target Genes by Co-occupancy of Transcription Factors, c-Myc and Mxi1 with Max in the Mouse Cell Line. Curr. Bioinform. 2020, 15, 581–588. [Google Scholar] [CrossRef]
- Cheng, S.; Li, D.; Zhang, R.-Z.; Zhu, J.; Wang, L.; Liu, Q.; Chen, R.-H.; Liu, X.-M. Characterization of Induced Pluripotent Stem Cells from Human Epidermal Melanocytes by Transduction with Two Combinations of Transcription Factors. Curr. Gene Ther. 2020, 19, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L.; et al. CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef]
- Zuo, Y.; Li, Y.; Chen, Y.; Li, G.; Yan, Z.; Yang, L. PseKRAAC: A flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics 2017, 33, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Liu, D.Y.; Yang, W.; Yang, L.; Zuo, Y.C. RaacLogo: A new sequence logo generator by using reduced amino acid clusters. Brief. Bioinform. 2021, 22, bbaa096. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, D.Y.; Li, Y.A.; Yang, S.Q.; Liang, Y.C.; Xing, Y.Q.; Zuo, Y.C. RaacFold: A webserver for 3D visualization and analysis of protein structure by using reduced amino acid alphabets. Nucleic Acids Res. 2022, 50, W633–W638. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.-H.; Zhao, X.; Wang, W. CD24, A Review of its Role in Tumor Diagnosis, Progression and Therapy. Curr. Gene Ther. 2020, 20, 109–126. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Yang, Y.H.; Ding, H.; Wang, D.; Chen, W.; Lin, H. Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief. Bioinform. 2021, 22, 526–535. [Google Scholar] [CrossRef]
- Hunt, C.; Montgomery, S.; Berkenpas, J.W.; Sigafoos, N.; Oakley, J.C.; Espinosa, J.; Justice, N.; Kishaba, K.; Hippe, K.; Si, D.; et al. Recent Progress of Machine Learning in Gene Therapy. Curr. Gene Ther. 2021, 22, 132–143. [Google Scholar] [CrossRef]
- Lv, H.; Dao, F.-Y.; Lin, H. DeepKla: An attention mechanism-based deep neural network for protein lysine lactylation site prediction. iMeta 2022, 1, e11. [Google Scholar] [CrossRef]
- Wei, L.; Liao, M.; Gao, Y.; Ji, R.; He, Z.; Zou, Q. Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wan, S.; Guo, J.; Wong, K.K.L. A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 2017, 83, 82–90. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Hasan, M.M.; Park, H.W.; Lee, K.W.; Manavalan, B. TACOS: A novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization. Brief. Bioinform. 2022, 23, bbac243. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Ning, L.; Ye, X.; Yang, Y.H.; Futamura, Y.; Sakurai, T.; Lin, H. iLoc-miRNA: Extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 2022, 23, bbac395. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Zhang, D.; Zhang, Z.M.; Liu, L.; Lin, H. DeepYY1: A deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. 2021, 22, bbaa356. [Google Scholar] [CrossRef]
- Basith, S.; Hasan, M.M.; Lee, G.; Wei, L.; Manavalan, B. Integrative machine learning framework for the identification of cell-specific enhancers from the human genome. Brief. Bioinform. 2021, 22, bbab252. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Patra, M.C. MLCPP 2.0: An Updated Cell-penetrating Peptides and Their Uptake Efficiency Predictor. J. Mol. Biol. 2022, 434, 167604. [Google Scholar] [CrossRef]
- Thi Phan, L.; Woo Park, H.; Pitti, T.; Madhavan, T.; Jeon, Y.J.; Manavalan, B. MLACP 2.0: An updated machine learning tool for anticancer peptide prediction. Comput. Struct. Biotechnol. J. 2022, 20, 4473–4480. [Google Scholar] [CrossRef]
- Zheng, L.; Huang, S.; Mu, N.; Zhang, H.; Zhang, J.; Chang, Y.; Yang, L.; Zuo, Y. RAACBook: A web server of reduced amino acid alphabet for sequence-dependent inference by using Chou’s five-step rule. Database 2019, 2019, baz131. [Google Scholar] [CrossRef] [Green Version]
- Alim, A.; Rafay, A.; Naseem, I. PoGB-pred: Prediction of Antifreeze Proteins Sequences Using Amino Acid Composition with Feature Selection Followed by a Sequential-based Ensemble Approach. Curr. Bioinform. 2021, 16, 446–456. [Google Scholar] [CrossRef]
- Yuan, S.S.; Gao, D.; Xie, X.Q.; Ma, C.Y.; Su, W.; Zhang, Z.Y.; Zheng, Y.; Ding, H. IBPred: A sequence-based predictor for identifying ion binding protein in phage. Comput. Struct. Biotechnol. J. 2022, 20, 4942–4951. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, H.; Liu, Y.; Li, J.; Wu, C.; Tang, H. Exosomal Non-Coding RNAs: New Insights into the Biology of Hepatocellular Carcinoma. Curr. Oncol. 2022, 29, 5383–5406. [Google Scholar] [CrossRef]
- Ye, Z.X.; Zhang, Y.X.; Liang, Y.B.; Lang, J.D.; Zhang, X.L.; Zang, G.L.; Yuan, D.W.; Tian, G.; Xiao, M.S.; Yang, J.L. Cervical Cancer Metastasis and Recurrence Risk Prediction Based on Deep Convolutional Neural Network. Curr. Bioinform. 2022, 17, 164–173. [Google Scholar]
- Liu, T.; Chen, J.M.; Zhang, D.; Zhang, Q.; Peng, B.; Xu, L.; Tang, H. ApoPred: Identification of Apolipoproteins and Their Subfamilies With Multifarious Features. Front. Cell. Dev. Biol. 2020, 8, 621144. [Google Scholar] [CrossRef]
- Zulfira, F.Z.; Suyanto, S.; Septiarini, A. Segmentation technique and dynamic ensemble selection to enhance glaucoma severity detection. Comput. Biol. Med. 2021, 139, 104951. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cao, R.Z.; Wang, W.; Liu, T.S.; Wang, L.M.; He, C.M. A two-step discriminated method to identify thermophilic proteins. Int. J. Biomath. 2017, 10, 1750050. [Google Scholar] [CrossRef]
- Zou, Y.; Wu, H.; Guo, X.; Peng, L.; Ding, Y.; Tang, J.; Guo, F. MK-FSVM-SVDD: A Multiple Kernel-based Fuzzy SVM Model for Predicting DNA-binding Proteins via Support Vector Data Description. Curr. Bioinform. 2021, 16, 274–283. [Google Scholar] [CrossRef]
- Wang, H.; Liang, P.F.; Zheng, L.; Long, C.S.; Li, H.S.; Zuo, Y.C. eHSCPr discriminating the cell identity involved in endothelial to hematopoietic transition. Bioinformatics 2021, 37, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Luo, Y.; Ren, X.; Wu, M.; He, X.; Peng, B.; Deng, K.; Yan, D.; Tang, H.; Lin, H. Risk Prediction of Diabetes: Big data mining with fusion of multifarious physical examination indicators. Inf. Fusion. 2021, 75, 140–149. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Fu, H.; Ruan, X.; Tang, X. DeepFusion-RBP: Using Deep Learning to Fuse Multiple Features to Identify RNA-binding Protein Sequences. Curr. Bioinform. 2021, 16, 1089–1100. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z.; Jiang, Y.; Mao, Z.; Wang, D.; Lin, H.; Xu, D. DM3Loc: Multi-label mRNA subcellular localization prediction and analysis based on multi-head self-attention mechanism. Nucleic Acids Res. 2021, 49, e46. [Google Scholar] [CrossRef]
- Lv, H.; Shi, L.; Berkenpas, J.W.; Dao, F.Y.; Zulfiqar, H.; Ding, H.; Zhang, Y.; Yang, L.; Cao, R. Application of artificial intelligence and machine learning for COVID-19 drug discovery and vaccine design. Brief. Bioinform. 2021, 22, bbab320. [Google Scholar] [CrossRef]
- Berahmand, K.; Nasiri, E.; Mohammadiani, R.P.; Li, Y. Spectral clustering on protein-protein interaction networks via constructing affinity matrix using attributed graph embedding. Comput. Biol. Med. 2021, 138, 104933. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Akbar, S.; Ghulam, A.; Maher, Z.A.; Unar, A.; Talpur, D.B. AFP-CMBPred: Computational identification of antifreeze proteins by extending consensus sequences into multi-blocks evolutionary information. Comput. Biol. Med. 2021, 139, 105006. [Google Scholar] [CrossRef]
- Yu, L.; Xia, M.; An, Q. A network embedding framework based on integrating multiplex network for drug combination prediction. Brief. Bioinform. 2022, 23, bbab364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jiang, H.; Gao, B.; Yang, W.; Wang, G. Identification of Diagnostic Markers for Breast Cancer Based on Differential Gene Expression and Pathway Network. Front. Cell. Dev. Biol. 2021, 9, 811585. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Gu, Y.; Zhu, J.; Ci, C.; Guo, Z.; Chen, C.; Wei, Y.; Lv, W.; Liu, H.; et al. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Mol. Oncol. 2018, 12, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhang, Y.; Wang, J.S.; Yuan, S.S.; Sun, Z.J.; Dao, F.Y.; Guan, Z.X.; Lin, H.; Deng, K.J. iRice-MS: An integrated XGBoost model for detecting multitype post-translational modification sites in rice. Brief. Bioinform. 2022, 23, bbab486. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. NPalmitoylDeep-pseaac: A predictor of N-Palmitoylation Sites in Proteins Using Deep Representations of Proteins and PseAAC via Modified 5-Steps Rule. Curr. Bioinform. 2021, 16, 294–305. [Google Scholar] [CrossRef]
- Ao, C.; Zou, Q.; Yu, L. NmRF: Identification of multispecies RNA 2′-O-methylation modification sites from RNA sequences. Brief. Bioinform. 2022, 23, bbab480. [Google Scholar] [CrossRef]
- Jin, Q.; Meng, Z.; Tuan, D.P.; Chen, Q.; Wei, L.; Su, R. DUNet: A deformable network for retinal vessel segmentation. Knowl. -Based Syst. 2019, 178, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. Nucleic Acids 2019, 16, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep-Resp-Forest: A deep forest model to predict anti-cancer drug response. Methods 2019, 166, 91–102. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Zeng, J.; Chen, J.; Su, R.; Guo, F. Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 2017, 83, 67–74. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Yu, L. A heterogeneous network embedding framework for predicting similarity-based drug-target interactions. Brief. Bioinform. 2021, 22, bbab275. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, H.; Yuan, S.S.; Huang, Q.L.; Sun, Z.J.; Dao, F.Y.; Yu, X.L.; Lin, H. Identification of cyclin protein using gradient boost decision tree algorithm. Comput. Struct. Biotechnol. J. 2021, 19, 4123–4131. [Google Scholar] [CrossRef]
- Zunair, H.; Ben Hamza, A. Sharp U-Net: Depthwise convolutional network for biomedical image segmentation. Comput. Biol. Med. 2021, 136, 104699. [Google Scholar] [CrossRef]
- Zhu, Q.; Fan, Y.; Pan, X. Fusing Multiple Biological Networks to Effectively Predict miRNA-disease Associations. Curr. Bioinform. 2021, 16, 371–384. [Google Scholar] [CrossRef]
- Yu, L.; Wang, M.; Yang, Y.; Xu, F.; Zhang, X.; Xie, F.; Gao, L.; Li, X. Predicting therapeutic drugs for hepatocellular carcinoma based on tissue-specific pathways. PLoS Comput. Biol. 2021, 17, e1008696. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Liu, J.; Wang, G. The stacking strategy-based hybrid framework for identifying non-coding RNAs. Brief. Bioinform. 2021, 22, bbab023. [Google Scholar] [CrossRef]
- Jiang, Q.H.; Wang, G.H.; Jin, S.L.; Li, Y.; Wang, Y.D. Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Linder, A.; Arnold, R.; Boyd, J.H.; Zindovic, M.; Zindovic, I.; Lange, A.; Paulsson, M.; Nyberg, P.; Russell, J.A.; Pritchard, D.; et al. Heparin-Binding Protein Measurement Improves the Prediction of Severe Infection With Organ Dysfunction in the Emergency Department. Crit. Care Med. 2015, 43, 2378–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parameters | SVM 1 | Parameters | RF |
|---|---|---|---|
| “kernel” | Linear, RBF, sigmoid, poly | “criterion” | Gini, entropy |
| “C” | 2x, x ∈ [−1, 15] | “max_depth” | [5, 150] |
| “gamma” | 2x, x ∈ [−14, 2] | “min_samples_split” | [2, 30] |
| “degree” | [1, 5] | “min_samples_leaf” | [5] |
| \ | \ | “max_leaf_nodes” | [100] |
| \ | \ | “ccp_alpha” | [0.001] |
| \ | \ | “n_estimators” | 10x, x ∈ [1, 3] |
| Feature Descriptor | Feature Name | F-Score | p-Value |
|---|---|---|---|
| AAC | S | 105.4221 | 4.9857 × 10−21 |
| C | 51.9136 | 6.0761 × 10−12 | |
| P | 39.1761 | 1.5583 × 10−9 | |
| V | 28.9828 | 1.6138 × 10−7 | |
| W | 18.6945 | 2.1764 × 10−5 | |
| DC | SS | 85.6575 | 7.6231 × 10−18 |
| PS | 64.2325 | 3.5827 × 10−14 | |
| SP | 63.8450 | 4.1972 × 10−14 | |
| PA | 39.7520 | 1.1720 × 10−9 | |
| QP | 28.6408 | 4.4865 × 10−8 | |
| DDE | SS | 87.9754 | 3.1559 × 10−18 |
| PS | 62.1955 | 8.2516 × 10−14 | |
| SP | 60.4646 | 1.6840 × 10−13 | |
| PA | 36.5852 | 4.9772 × 10−9 | |
| FC | 29.5543 | 1.2376 × 10−7 | |
| CTD | solventaccess.G3 | 93.4288 | 4.0577 × 10−19 |
| hydrophobicity_ARGP820101.G2 | 83.7918 | 1.5572 × 10−17 | |
| polarity.G3 | 80.3504 | 5.8770 × 10−17 | |
| hydrophobicity_ZIMJ680101.G1 | 73.3974 | 8.9800 × 10−16 | |
| secondarystruct.G1 | 69.7518 | 3.8413 × 10−15 |
| Algorithm | Feature | Sn (%) | Sp (%) | MCC | OA (%) |
|---|---|---|---|---|---|
| SVM | AAC | 98.0 | 88.0 | 0.864 | 93.0 |
| DC | 92.0 | 98.0 | 0.902 | 95.0 | |
| DDE | 96.0 | 94.0 | 0.900 | 95.0 | |
| CTD | 94.0 | 94.0 | 0.880 | 94.0 | |
| RF | AAC | 90.0 | 86.0 | 0.761 | 88.0 |
| DC | 96.0 | 94.0 | 0.900 | 95.0 | |
| DDE | 96.0 | 86.0 | 0.824 | 91.0 | |
| CTD | 88.0 | 98.0 | 0.864 | 93.0 |
| Parameters | Value |
|---|---|
| “kernel” | RBF |
| “C” | 4.59479341998814 |
| “gamma” | 0.07982260524725553 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Yuan, S.-S.; Li, J.; Huang, C.-B.; Lin, H.; Liao, B. A First Computational Frame for Recognizing Heparin-Binding Protein. Diagnostics 2023, 13, 2465. https://doi.org/10.3390/diagnostics13142465
Zhu W, Yuan S-S, Li J, Huang C-B, Lin H, Liao B. A First Computational Frame for Recognizing Heparin-Binding Protein. Diagnostics. 2023; 13(14):2465. https://doi.org/10.3390/diagnostics13142465
Chicago/Turabian StyleZhu, Wen, Shi-Shi Yuan, Jian Li, Cheng-Bing Huang, Hao Lin, and Bo Liao. 2023. "A First Computational Frame for Recognizing Heparin-Binding Protein" Diagnostics 13, no. 14: 2465. https://doi.org/10.3390/diagnostics13142465
APA StyleZhu, W., Yuan, S. -S., Li, J., Huang, C. -B., Lin, H., & Liao, B. (2023). A First Computational Frame for Recognizing Heparin-Binding Protein. Diagnostics, 13(14), 2465. https://doi.org/10.3390/diagnostics13142465