
Evaluation and Analysis of Absence of Homozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis
Abstract
:1. Introduction
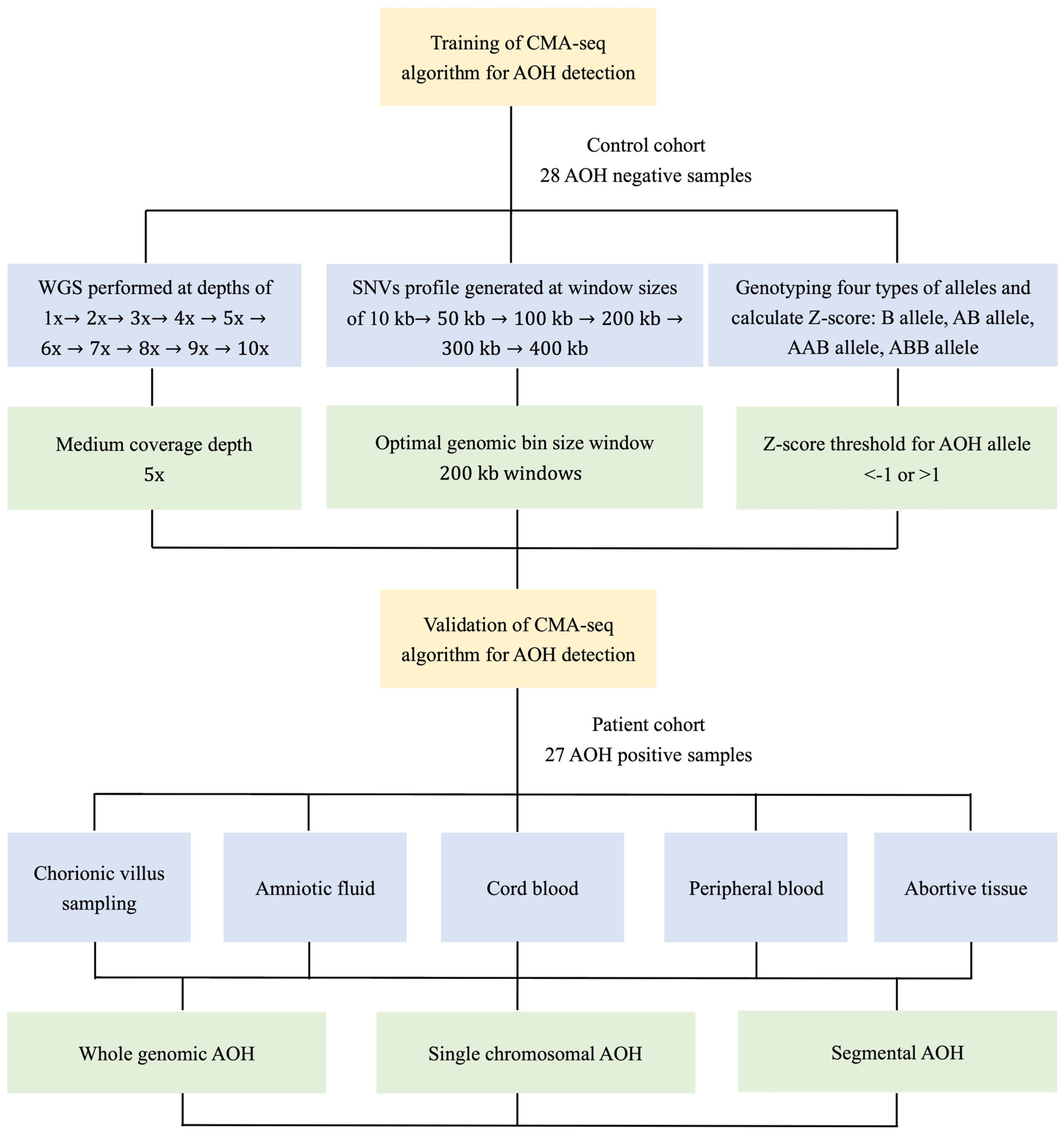
2. Materials and Methods
2.1. Study Design and Sample Preparation
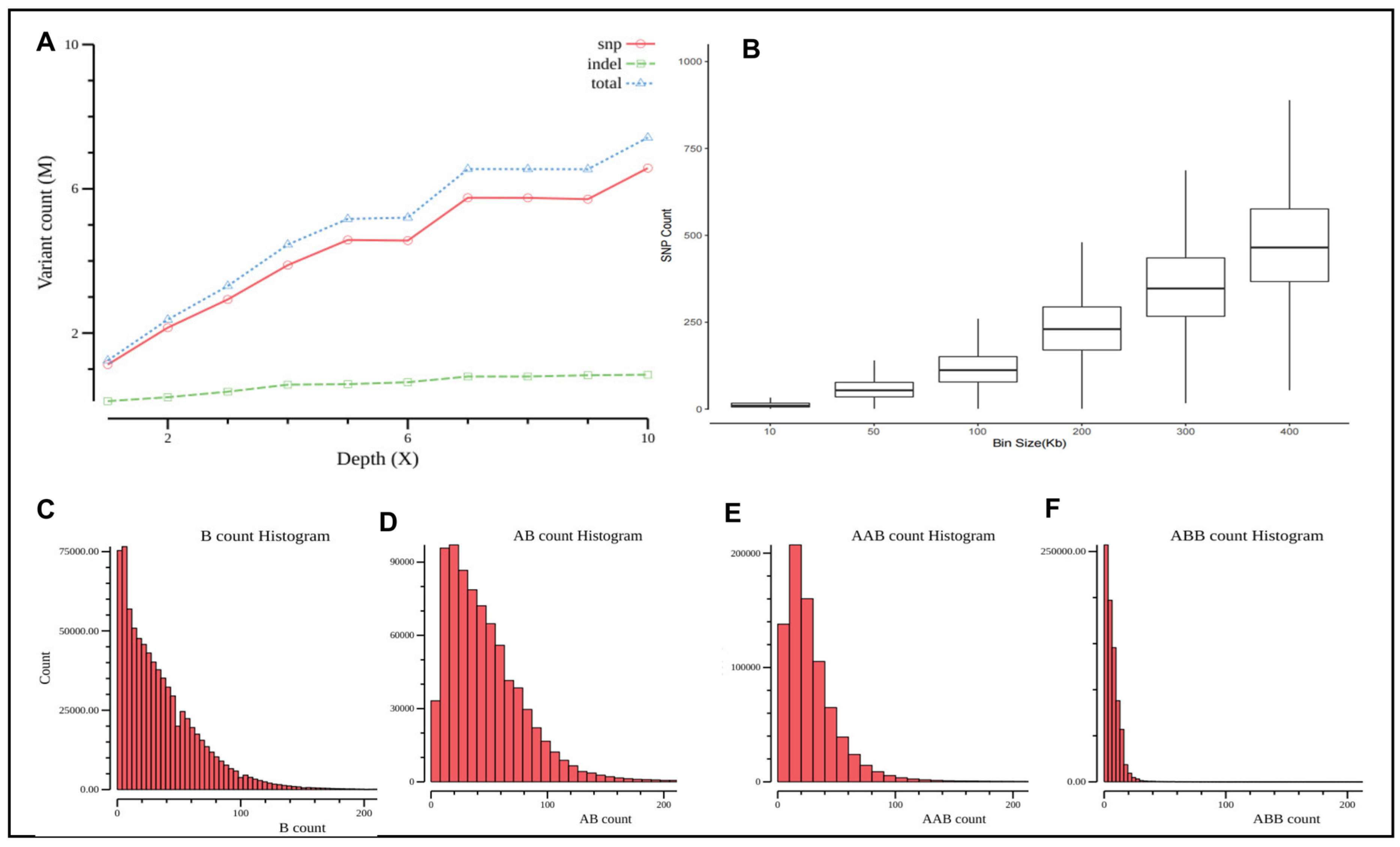
2.2. Whole Genome Sequencing at Different Coverage Depths
2.3. Bioinformatics Analysis of AOH Detection
2.4. Validation of AOH Detection by CMA/SNP Array
3. Results
3.1. Assessment of Minimum Sequencing Coverage for AOH Detection
3.2. Performance of Detection of AOH for Prenatal Samples with CMA-seq
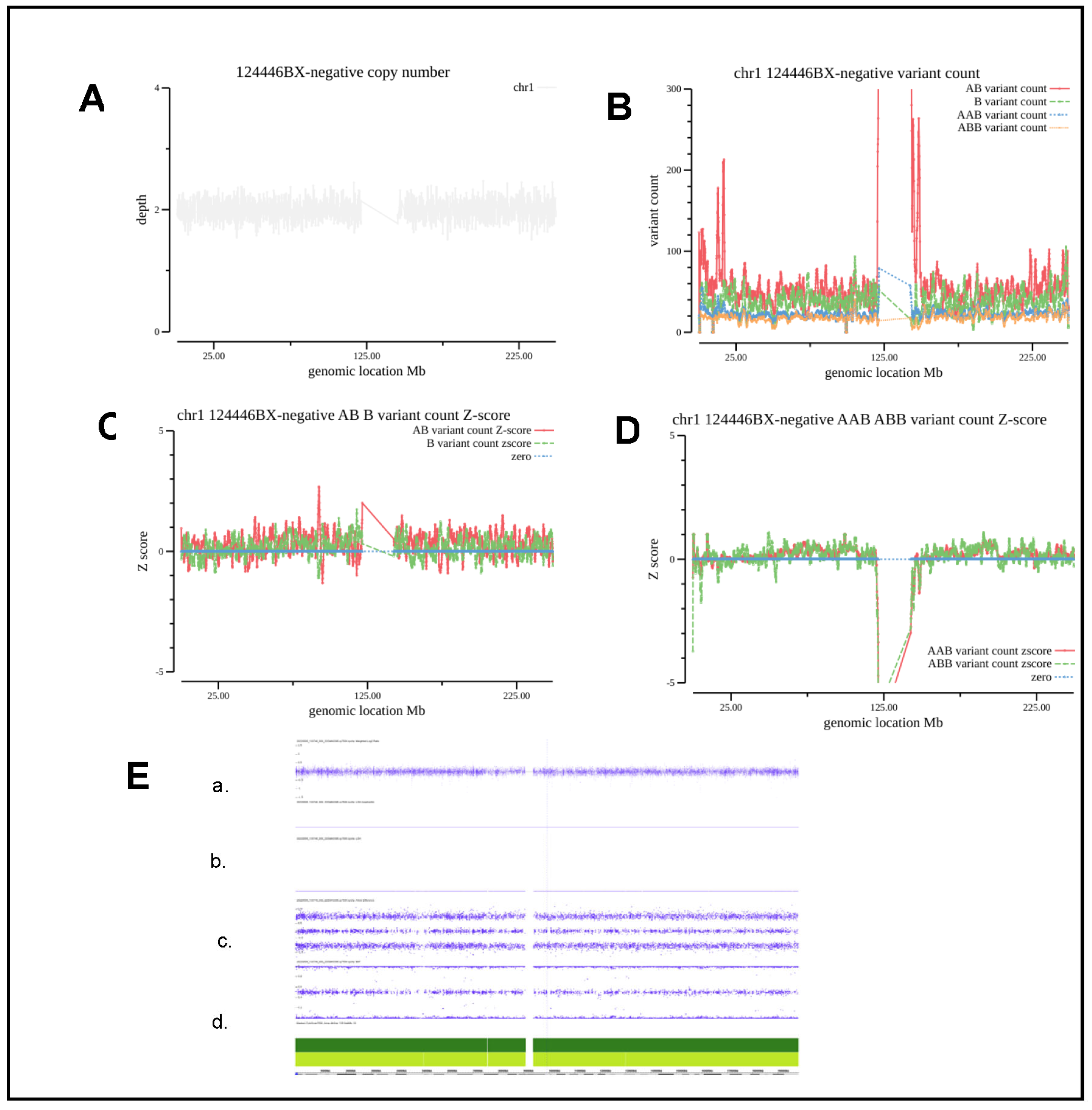
3.3. Evaluation and Analysis of Normal Sample and Detection of Chromosomal Level AOH
3.4. Evaluation and Analysis of Whole Genome Level AOH
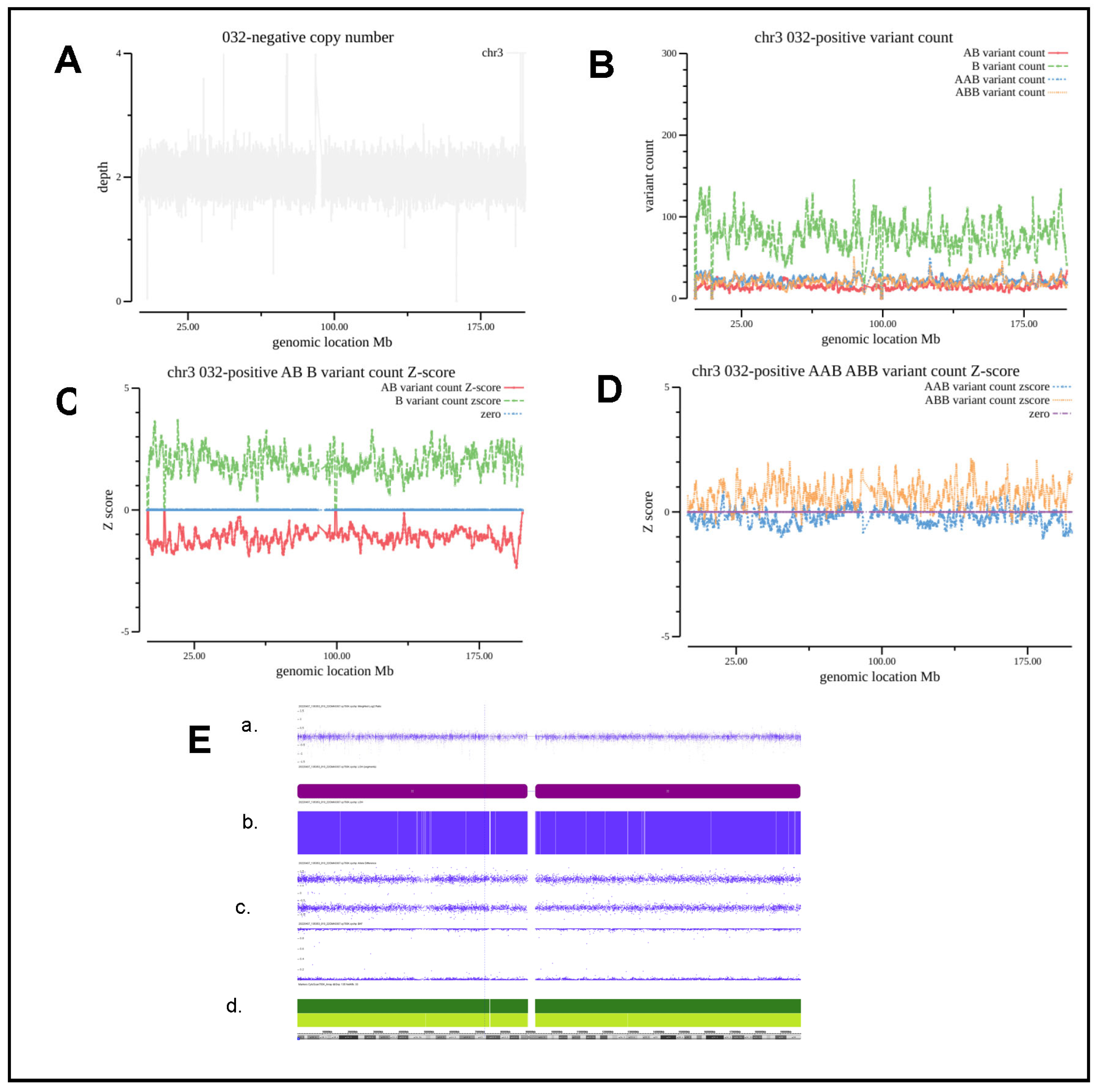
3.5. Evaluation and Analysis of Segmental AOH
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Bi, W. Novel applications of array comparative genomic hybridization in molecular diagnostics. Expert Rev. Mol. Diagn. 2018, 18, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Huang, J.; Greshock, J.; Watt, S.; Butler, A.; West, S.; Grigorova, M.; Jones, K.W.; Wei, W.; Stratton, M.R.; et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004, 14, 287–295. [Google Scholar] [CrossRef]
- Zhao, X.; Li, C.; Paez, J.G.; Chin, K.; Jänne, P.A.; Chen, T.H.; Girard, L.; Minna, J.; Christiani, D.; Leo, C.; et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004, 64, 3060–3071. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 581: The use of chromosomal microarray analysis in prenatal diagnosis. Obstet. Gynecol. 2013, 122, 1374–1377. [Google Scholar]
- Dong, Z.; Zhang, J.; Hu, P.; Chen, H.; Xu, J.; Tian, Q.; Meng, L.; Ye, Y.; Wang, J.; Zhang, M.; et al. Low-pass whole-genome sequencing in clinical cytogenetics: A validated approach. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 940–948. [Google Scholar] [CrossRef]
- Nakka, P.; Pattillo Smith, S.; O’Donnell-Luria, A.H.; McManus, K.F.; Mountain, J.L.; Ramachandran, S.; Sathirapongsasuti, J.F. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 2019, 105, 921–932. [Google Scholar] [CrossRef]
- Wang, L.; Liu, P.; Bi, W.; Sim, T.; Wang, X.; Walkiewicz, M.; Leduc, M.S.; Meng, L.; Xia, F.; Eng, C.M.; et al. Contribution of uniparental disomy in a clinical trio exome cohort of 2675 patients. Mol. Genet. Genom. Med. 2021, 9, e1792. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Ahn, J.W.; Bagnall, R.D.; Baralle, D.; Barton, S.; Campbell, C.; Downes, K.; Ellard, S.; Duff-Farrier, C.; FitzPatrick, D.R.; et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022, 14, 73. [Google Scholar] [CrossRef]
- Liang, D.; Peng, Y.; Lv, W.; Deng, L.; Zhang, Y.; Li, H.; Yang, P.; Zhang, J.; Song, Z.; Xu, G.; et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J. Mol. Diagn. JMD 2014, 16, 519–526. [Google Scholar] [CrossRef]
- Hayes, J.L.; Tzika, A.; Thygesen, H.; Berri, S.; Wood, H.M.; Hewitt, S.; Pendlebury, M.; Coates, A.; Willoughby, L.; Watson, C.M.; et al. Diagnosis of copy number variation by Illumina next generation sequencing is comparable in performance to oligonucleotide array comparative genomic hybridisation. Genomics 2013, 102, 174–181. [Google Scholar] [CrossRef]
- Cohen, K.; Tzika, A.; Wood, H.; Berri, S.; Roberts, P.; Mason, G.; Sheridan, E. Diagnosis of fetal submicroscopic chromosomal abnormalities in failed array CGH samples: Copy number by sequencing as an alternative to microarrays for invasive fetal testing. Ultrasound Obs. Gynecol 2015, 45, 394–401. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Xie, W.; Vogel, I.; Choy, K.W.; Chen, F.; Christensen, R.; Zhang, C.; Ge, H.; Jiang, H.; et al. PSCC: Sensitive and reliable population-scale copy number variation detection method based on low coverage sequencing. PLoS ONE 2014, 9, e85096. [Google Scholar] [CrossRef]
- Dong, Z.; Xie, W.; Chen, H.; Xu, J.; Wang, H.; Li, Y.; Wang, J.; Chen, F.; Choy, K.W.; Jiang, H. Copy-Number Variants Detection by Low-Pass Whole-Genome Sequencing. Curr. Protoc. Hum. Genet. 2017, 94, 8.17.11–18.17.16. [Google Scholar] [CrossRef]
- Qi, Q.; Jiang, Y.; Zhou, X.; Meng, H.; Hao, N.; Chang, J.; Bai, J.; Wang, C.; Wang, M.; Guo, J.; et al. Simultaneous Detection of CNVs and SNVs Improves the Diagnostic Yield of Fetuses with Ultrasound Anomalies and Normal Karyotypes. Genes 2020, 11, 1397. [Google Scholar] [CrossRef]
- Liu, X.; Liu, S.; Wang, H.; Hu, T. Potentials and challenges of chromosomal microarray analysis in prenatal diagnosis. Front. Genet. 2022, 13, 938183. [Google Scholar] [CrossRef]
- Wang, H.; Dong, Z.; Zhang, R.; Chau, M.H.K.; Yang, Z.; Tsang, K.Y.C.; Wong, H.K.; Gui, B.; Meng, Z.; Xiao, K.; et al. Low-pass genome sequencing versus chromosomal microarray analysis: Implementation in prenatal diagnosis. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 500–510. [Google Scholar] [CrossRef]
- Chaubey, A.; Shenoy, S.; Mathur, A.; Ma, Z.; Valencia, C.A.; Reddy Nallamilli, B.R.; Szekeres, E., Jr.; Stansberry, L.; Liu, R.; Hegde, M.R. Low-Pass Genome Sequencing: Validation and Diagnostic Utility from 409 Clinical Cases of Low-Pass Genome Sequencing for the Detection of Copy Number Variants to Replace Constitutional Microarray. J. Mol. Diagn. JMD 2020, 22, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lin, S.; Zhao, X.; Bennett, M.F.; Brown, N.J.; Wallis, M.; Gao, X.; Sun, L.; Wu, J.; Vedururu, R.; et al. Mosaicism in tuberous sclerosis complex: Lowering the threshold for clinical reporting. Hum. Mutat. 2022, 43, 1956–1969. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chau, M.H.K.; Zhang, Y.; Yang, Z.; Shi, M.; Wah, Y.M.; Kwok, Y.K.; Leung, T.Y.; Morton, C.C.; Choy, K.W. Low-pass genome sequencing-based detection of absence of heterozygosity: Validation in clinical cytogenetics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kishikawa, T.; Momozawa, Y.; Ozeki, T.; Mushiroda, T.; Inohara, H.; Kamatani, Y.; Kubo, M.; Okada, Y. Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Sci. Rep. 2019, 9, 1784. [Google Scholar] [CrossRef]
- Benjelloun, B.; Boyer, F.; Streeter, I.; Zamani, W.; Engelen, S.; Alberti, A.; Alberto, F.J.; BenBati, M.; Ibnelbachyr, M.; Chentouf, M.; et al. An evaluation of sequencing coverage and genotyping strategies to assess neutral and adaptive diversity. Mol. Ecol. Resour. 2019, 19, 1497–1515. [Google Scholar] [CrossRef]
- Lu, Y.H.; Wang, B.H.; Xia, W.; Mo, X.B.; Wu, L.F.; Zhu, X.W.; He, P.; Xie, F.F.; Lu, X.; Deng, F.Y.; et al. The distribution and functional relevance analysis of runs of homozygosity (ROHs) in Chinese Han female population. Mol. Genet. Genom. 2018, 293, 197–206. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Xie, C.; Tammi, M.T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinform. 2009, 10, 80. [Google Scholar] [CrossRef]
- Chau, M.H.K.; Qian, J.; Chen, Z.; Li, Y.; Zheng, Y.; Tse, W.T.; Kwok, Y.K.; Leung, T.Y.; Dong, Z.; Choy, K.W. Trio-Based Low-Pass Genome Sequencing Reveals Characteristics and Significance of Rare Copy Number Variants in Prenatal Diagnosis. Front. Genet. 2021, 12, 742325. [Google Scholar] [CrossRef]
- Sparks, T.N.; Dugoff, L. How to choose a test for prenatal genetic diagnosis: A practical overview. Am. J. Obstet. Gynecol. 2022, 228, 178–186. [Google Scholar] [CrossRef]
- Dugoff, L.; Norton, M.E.; Kuller, J.A.; Medicine, S.f.M.-F. The use of chromosomal microarray for prenatal diagnosis. Am. J. Obstet. Gynecol. 2016, 215, B2–B9. [Google Scholar] [CrossRef]
- Chau, M.H.K.; Wang, H.; Lai, Y.; Zhang, Y.; Xu, F.; Tang, Y.; Wang, Y.; Chen, Z.; Leung, T.Y.; Chung, J.P.W.; et al. Low-pass genome sequencing: A validated method in clinical cytogenetics. Hum. Genet. 2020, 139, 1403–1415. [Google Scholar] [CrossRef]
- Zhu, X.; Li, J.; Ru, T.; Wang, Y.; Xu, Y.; Yang, Y.; Wu, X.; Cram, D.S.; Hu, Y. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat. Diagn. 2016, 36, 321–327. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, X.; Hu, J.; He, G.; Wang, J.; Zhu, Y.; Zhu, B. Investigation on combined copy number variation sequencing and cytogenetic karyotyping for prenatal diagnosis. BMC Pregnancy Childbirth 2021, 21, 496. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samle ID | Sample Types | Clinical Indication(s) | CMA | GS | Result of Comparison | Classification | Diseases | ||
|---|---|---|---|---|---|---|---|---|---|
| ‘001 | amniotic fluid | Advanced Maternal Age, The couple had a child with chromosomal disease (46, X, iY(p10)) | 7q21.11q21.3(80,832,371–94,133,800)×2 hmz | 13.3 | 7q21.11q21.3(81,000,000–94,100,000), ×2 hmz | 13.1 | Consistently | VUS | Silver–Russell syndrome |
| ‘002 | amniotic fluid | advanced maternal age, abnormal NIPT result (Z2 = 9.9) | 2p25.3p24.3(50,814–13,311,915)×2 hmz | 13 | 2p25.3p24.3(200,000–13,100,000), ×2 hmz | 12.9 | Consistently | VUS | |
| 2q32.3q36.3(192,341,274–230,205,775)×2 hmz | 37.9 | 2q33.2q36.3(204,100,000–230,100,000), ×2 hmz | 26 | Consistently | VUS | ||||
| 2p21p11.2(45,974,855–87,053,152)×2 hmz | 41.1 | 2p21p13.2(45,700,000–72,400,000), ×2 hmz; 2p13.1p11.2(75,000,000–89,000,000), ×2 hmz | 26.7 14 | Consistently | VUS | ||||
| 2q11.1q12.3(95,550,958–109,626,929)×2 hmz | 14 | 2q11.2q12.3(98,700,000–108,200,000), ×2hmz | 9.5 | Consistently | VUS | ||||
| ‘027 | peripheral blood | parental validation for abnormal fetal CMA result. The fetus had an ultrasound abnormality of omphalocele, and the CMA indicated a 539 kb duplication on chromosome 16p11.2, which was associated with 16p11.2 duplication syndrome, as well as a 1.7 Mb duplication on chromosome Xp22.31 of unknow significance. | Xp22.31(6,455,152–8,135,568)×4 | Xp22.31(6,455,152–8,135,568)x3.88 | Consistently | VUS | |||
| 4q25q28.3(111,718,067–137,498,491)×2 hmz | 25 | 4q25q28.3(111,800,000–137,400,000), ×2hmz | 25.6 | Consistently | VUS | ||||
| 6q14.1q21(83,539,461–113,232,989)×2 hmz | 29 | 6q14.1q21(83,500,000–113,200,000), ×2hmz | 29.7 | Consistently | VUS | transient neonatal diabetes mellitus | |||
| 7q31.1q33(112,161,869–136,999,883)×2 hmz | 24 | 7q31.1q33(112,300,000–136,400,000), ×2hmz | 24.1 | Consistently | VUS | Silver–Russell syndrome | |||
| Xp22.31p21.2(8,485,023–31,282,369)×2 hmz, | 23 | Xp22.31p22.2(7,900,000–10,200,000), ×2 hmz; Xp22.2(11,500,000–14,300,000), ×2 hmz; Xp22.2p22.13(15,300,000–17,800,000), ×2 hmz; Xp22.12p21.3(20,400,000–27,700,000), ×2 hmz; Xp21.3p21.2(28,500,000–31,200,000), ×2 hmz; Xp11.21(49,00,000–57,600,000), ×2 hmz; Xq21.1(80,100,000–82,400,000), ×2 hmz | 2.3 2.8 2.5 7.3 2.7 2.7 2.3 | Consistently | VUS | ||||
| Xq23q25(114,567,797–128,598,791)×2 hmz | 14 | Xq24q25(117,900,000–128,600,000), ×2 hmz | 10.7 | Consistently | VUS | ||||
| 7q36.2q36.3(154,400,000–158,800,000), ×2 hmz | 4.4 | Additional findings | VUS | ||||||
| 20p13p12.3(100,000–5,800,000), ×2 hmz | 5.7 | Additional findings | P | Pseudohypoparathyroidism type 1B | |||||
| ‘030 | chorionic villus sampling | The ultrasound indicated fetal abnormality (NT = 3.3 mm) | 2p12p11.2(75,632,552–87,053,152)×2 hmz, | 11.4 | 2p12p11.2(75,600,000–87,100,000), ×2 hmz | 11.5 | Consistently | VUS | |
| 15q25.1q26.1(79,625,730–94,285,672)×2 hmz | 14.7 | 15q25.1q26.1(78,900,000–94,200,000), ×2 hmz | 15.3 | Consistently | VUS | Prader Willi syndrome, Angelman syndrome | |||
| ‘031 | peripheral blood | systemic scleroderma | 1p13.3p11.2(107,624,475_121,339,317)×2 hmz, | 13.7 | 1p13.3p12(107,500,000–120,500,000), ×2 hmz | 13 | Consistently | ||
| 1q21.1q32.2(144,077,594_207,345,376)×2 hmz, | 63.3 | 1q21.3q22(151,200,000–155,300,000), ×2 hmz; 1q22q32.1(155,900,000–206,000,000), ×2 hmz | 4.1 50.1 | Consistently | VUS | ||||
| 7q11.22q21.11(71,236,774_83,356,842)×2 hmz, | 12 | 7q11.23q21.11(75,200,000–83,400,000), ×2 hmz | 8.2 | Consistently | VUS | Silver–Russell syndrome | |||
| 10p14p12.1(7,588,527_29,290,352)×2 hmz | 21.7 | 10p14p12.31(7,700,000–21,600,000), ×2 hmz; 10p12.31p12.1(22,200,000–29,600,000), ×2 hmz | 13.9 7.4 | Consistently | VUS | ||||
| Xp22.33p22.31(2,100,000–8,500,000), ×2 hmz | 6.4 | Additional findings | VUS | ||||||
| Xp11.21(54,900,000–57,600,000), ×2 hmz | 2.7 | Additional findings | VUS | ||||||
| Xq13.3q21.1(74,400,000–77,000,000), ×2 hmz | 2.6 | Additional findings | VUS | ||||||
| Xq24q26.3(119,500,000–136,400,000), ×2 hmz | 16.9 | Additional findings | VUS | ||||||
| 7p14.1(40,390,001–40,530,000), ×1 | ~140 kb, deletion | Additional findings | LP | Glutaric aciduria type III | |||||
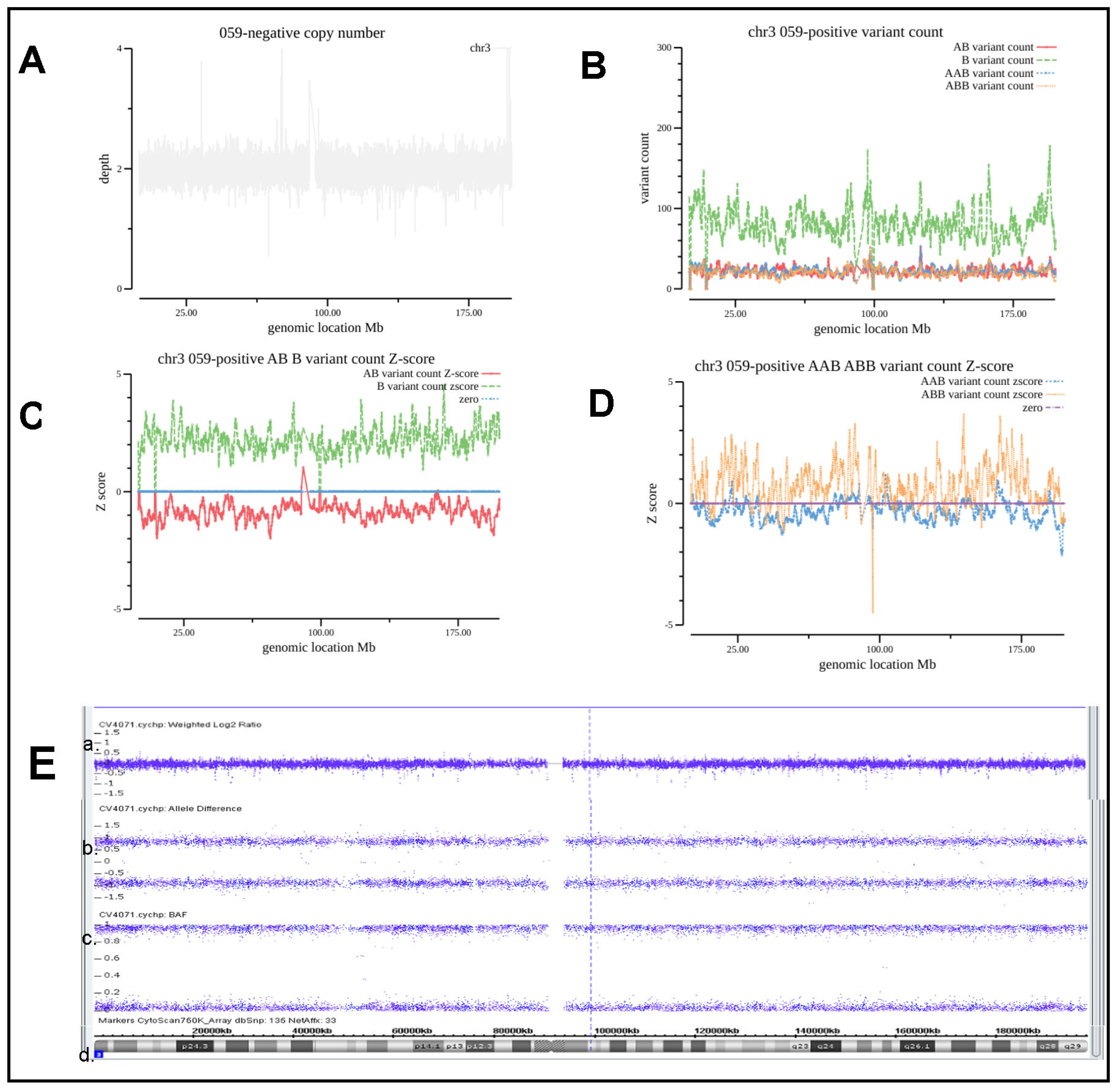
| ‘032 | amniotic fluid | abnormal NIPT result (Z3 = 18.49) | arr(3)×2 hmz | 3UPD | chr3 | 3UPD | Consistently | VUS | |
| ‘033 | amniotic fluid | advanced maternal age | 18q21.2q22.3(49,750,952_71,592,671)×2 hmz | 21.8 | 18q21.2q22.3(49,900,000–71,700,000), ×2 hmz | 21.8 | Consistently | VUS | |
| ‘034 | peripheral blood | parental validation for fetal CMA result. The fetus had abnormal NIPT result (Z18 = −7.11) and the CMA indicated a pathogenic variant of 15Mb deletion on chromosome 18q22.1q23, as well as a 6.4 Mb duplication on chromosome 1q43q44 of unknow significance. | 1q43(241,124,639_242,772,875)x1, | 1.65 | 1q43(241,110,001–242,770,000), ×2 hmz | 1.66, deletion | Consistently | P | |
| 2q32.3q34(192,406,884_209,298,633)×2 hmz | 16.9 | 2q32.3q34(192,700,000–209,300,000), ×2 hmz | 16.6 | Consistently | VUS | ||||
| 2q31.1q31.2(176,400,000–178,600,000), ×2 hmz | 2.2 | Additional findings | VUS | ||||||
| 18q22.1(63,100,001–63,680,000), ×2 hmz | ~580 kb, deletion | Additional findings | VUS | ||||||
| ‘036 | amniotic fluid | NT thickening | 13q11q34(19,450,957–115,095,705)×2 hmz | 95.6 | 13q11q34(19,300,000–115,100,000), ×2 hmz | 95.8 | Consistently | VUS | |
| ‘048 | amniotic fluid | fetal growth restriction | 8q12.1q13.3(58,247,980–71,953,755)×2 hmz | 13.7 | 8q12.1q13.3(58,400,000–73,000,000), ×2 hmz | 14.6 | Consistently | VUS | |
| ‘049 | amniotic fluid | nasal bone absence | 3p23p21.31(31,569,962–44,915,992)×2 hmz, | 13.3 | 3p23p22.2(31,700,000–44,400,000), ×2 hmz | 13.6 | Consistently | VUS | |
| (21)×3 | trisomy 21 | chr21×3 | trisomy 21 | Consistently | P | Down syndrome | |||
| ‘053 | peripheral blood | parental validation for abnormal fetal CMA result. The pregnancy women underwent chorionic villus sampling because the first child of the couple had phenylketonuria. The CMA indicated a 208 kb deletion on chromosome 4q25 of unknown significance. | 4q21.23q26(85,439,001–116,726,668)×2 hmz | 31.2 | 4q21.23q26(85,400,000–116,500,000), ×2 hmz | 31.1 | Consistently | VUS | |
| 8q22.2q24.21(101,423,593–128,671,420)×2 hmz | 27.2 | 8q22.3q24.21(101,700,000–129,100,000), ×2 hmz | 27.4 | Consistently | VUS | ||||
| 12p13.31p12.1(8,146,161–25,668,635)×2 hmz | 17.5 | 12p13.31p12.1(8,200,000–25,700,000), ×2 hmz | 17.5 | Consistently | VUS | ||||
| 4q28.3q31.1(138,700,000–141,000,000), ×2 hmz | 2.3 | Additional findings | VUS | ||||||
| 8q12.3q13.1(64,500,000–66,600,000), ×2 hmz | 2.1 | Additional findings | VUS | ||||||
| ‘054 | chorionic villus sampling | twin molar pregnancy | 19p13.3p11(260,911–24,462,369)×2 hmz, | 19 UPD | 19p13.3p12(400,000–20,900,000), ×2 hmz | 20.5 | Consistently | VUS | |
| 19q11q13.43(28,274,010–58,955,556)×2 hmz | 19p12(21,900,000–24,400,000), ×2 hmz | 2.5 | Consistently | VUS | |||||
| 19q11q13.2(28,500,000–42,500,000), ×2 hmz | 14 | Consistently | VUS | ||||||
| 19q13.31q13.43(43,500,000–58,900,000), ×2 hmz | 15.4 | Consistently | VUS | ||||||
| ‘055 | chorionic villus sampling | omphalocele | 13q12.11q13.3(22,951,566–38,289,937)×2 hmz, | 15.3 | 13q12.11q13.3(23,100,000–38,300,000), ×2 hmz | 15.2 | Consistently | VUS | |
| 18p11.32q23(136,227–78,013,728)×3 | trisomy 18 | chr18×3 | trisomy 18 | Consistently | VUS | ||||
| ‘056 | peripheral blood | intrauterine demise | Xq22.1q26.3(99,176,543–135,657,966)×2 hmz, | 36.4 | Xq22.1q22.3(99,600,000–104,300,000), ×2 hmz; Xq23q24(111,600,000–117,100,000), ×2 hmz; Xq24q25chrX:117,400,000–123,900,000, ×2 hmz; Xq25q26.3(126,700,000–134,800,000), ×2 hmz | 4.1 5.3 6.4 6.4 | Consistently | VUS | |
| 2q21.3q24.3(135,219,538–164,635,629)×2 hmz, | 29.4 | 2q21.2q24.1(134,700,000–158,200,000), ×2 hmz; 2q24.1q24.3(158,600,000–164,600,000), ×2 hmz | 23.5 6 | Consistently | VUS | ||||
| 7p22.3p21.2(50,943–14,472,953)×2 hmz, | 14.4 | 7p22.3p21.2(500,000–14,600,000), ×2 hmz | 14.1 | Consistently | VUS | Silver–Russell syndrome | |||
| 12p13.33p11.21(257,935–31,063,131)×2 hmz, | 30.8 | 12p13.33p11.21(400,000–31,300,000), ×2 hmz | 30.9 | Consistently | VUS | ||||
| 12q12q14.2(42,805,029–63,265,817)×2 hmz | 20.4 | 12q12q14.2(42,600,000–64,800,000), ×2 hmz | 22.2 | Consistently | VUS | ||||
| 7q31.32q31.33(122,600,000–125,200,000), ×2 hmz | 2.6 | Additional findings | VUS | ||||||
| 2p15p14(62,900,000–65,000,000), ×2 hmz | 2.1 | Additional findings | VUS | ||||||
| 2p12p11.2(81,700,000–84,000,000), ×2 hmz | 2.3 | Additional findings | VUS | ||||||
| ‘057 | Placenta | twin molar pregnancy | 19p13.3p11(260,911–24,462,369)×2 hmz, | 19 UPD | 19p13.3p13.11(400,000–19,500,000), ×2 hmz, | 19.1 | Consistently | VUS | |
| 19q11q13.43(28,274,010–58,955,556)×2 hmz | 19q11q13.43(28,500,000–59,100,000), ×2 hmz | 30.6 | Consistently | VUS | |||||
| ‘058 | amniotic fluid | congenital diaphragmatic hernia | 6p25.3q27(203,877–170,896,644)×2 hmz | 6 UPD | 6p25.3p11.1(200,000–58,900,000), ×2 hmz | 58.7 | Consistently | VUS | transient neonatal diabetes mellitus |
| 6q11.1q27(62,100,000–171,200,000), ×2 hmz | 109.1 | Consistently | VUS | transient neonatal diabetes mellitus | |||||
| ‘059 | chorionic villus sampling | twin molar pregnancy | arr(1–22,X)×2 hmz | Genome-wide | chr(1–22,X)×2 hmz | Consistently | P? | ||
| ‘061 | amniotic fluid | advanced maternal age | 15q22.2q25.3(61,837,131–86,568,353)×2 hmz | 24.7 | 15q22.2q25.2(61,900,000–82,600,000), ×2 hmz | 20.7 | Consistently | VUS | Prader Willi syndrome, Angelman syndrome |
| 15q25.2q25.3(83,300,000–87,300,000), ×2 hmz | 4 | Consistently | VUS | Prader Willi syndrome, Angelman syndrome | |||||
| Xp22.13(18,800,001–19,180,000), x3 | ~380 kb, duplication | Additional findings | VUS | ||||||
| ‘062 | peripheral blood | parental validation for fetal CMA result. The fetus had ultrasound abnormality of hyperechogenic kidneys and the CMA indicated a 1.4 Mb deletion on chromosome 17q12, which was associated with 17q12 recurrent deletion syndrome. | 6q15q21(91,258,687–106,596,685)×2 hmz | 15.3 | 6q15q21(91,400,000–106,500,000), ×2 hmz | 15.1 | Consistently | VUS | transient neonatal diabetes mellitus |
| ‘064 | chorionic villus sampling | megacystis, single umbilical artery | 20p12.3p11.23(9,051,988–20,268,154)×2 hmz | 20p12.3p11.23(9,000,000–20,300,000), ×2 hmz | 11.3 | Consistently | VUS | Pseudohypoparathyroidism type 1B | |
| ‘065 | cord blood | amniotic fluid sample revealing karotype of 47,XXY/46,XY | arr(X)×2 hmz,(Y)×1 | XXY, XUPD | arr(X)×2 hmz,(Y)×1 | X UPD | Consistently | VUS | |
| ‘066 | amniotic fluid | abnormal NIPT result | 3p24.1p14.2(30,639,805–63,601,962)×2 hmz, | 32.9 | 3p24.1p21.31(30,800,000–48,200,000), ×2 hmz; 3p21.31p14.2(50,100,000–63,500,000), ×2 hmz | 17.4 13.4 | Consistently | VUS | |
| 5q11.2q13.2(54,259,794–68,826,246)×2 hmz, | 14.5 | 5q11.2q13.2(54,100,000–71,200,000), ×2 hmz | 17.1 | Consistently | VUS | ||||
| 9p23p21.2(11,072,446–25,606,655)×2 hmz, | 14.5 | 9p23p21.3(11,200,000–25,500,000), ×2 hmz | 14.3 | Consistently | VUS | ||||
| 18q21.1q21.32(44,375,110–57,719,351)×2 hmz | 13.3 | 18q21.1q21.32(44,400,000–57,700,000), ×2 hmz | 13.3 | Consistently | VUS | ||||
| 3p26.1p25.2(6,800,000–12,300,000), ×2 hmz | 5.5 | Additional findings | VUS | ||||||
| VUS | |||||||||
| ‘067 | peripheral blood | parental validation for fetal CMA result. The fetus had ultrasound abnormality of congenital diaphragmatic hernia and the CMA indicated a 335 kb duplication on chromosome 7q36.1 of unknow significance. | 7q36.1(151,842,964–152,178,027)×3 | 335 kb, duplication | 7q36.1(151,840,001–152,070,000)×3 | ~230 kb, duplication | Consistently | VUS | |
| 12q23.1q24.13(98,658,599–114,095,220)×2 hmz | 15.4 | 12q23.1q24.11(99,500,000–110,300,000), ×2 hmz | 10.8 | Consistently | VUS | ||||
| 16p13.3(5,910,001–6,260,000), ×1 | ~350 kb, deletion | Additional findings | VUS | ||||||
| ‘068 | peripheral blood | parental validation for fetal CMA result. The fetus had ultrasound abnormality of cleft lip and palate and the CMA indicated a 14.9 Mb deletion on chromosome 18p11.32p11.21, which was associated with Chromosome 18p deletion syndrome. | arr[hg19] 9q31.1q33.3(105,122,292–129,460,914)×2 hmz, | 24.3 | 9q31.1q33.3(104,800,000–129,400,000), ×2 hmz | 24.6 | Consistently | VUS | Temple syndrome, Kagami-Ogata syndrome |
| 12p13.2p12.1(10,121,410–24,518,044)×2 hmz | 14.3 | 12p13.2p12.1(10,200,000–24,700,000), ×2 hmz | 14.5 | Consistently | VUS | ||||
| 14q24.3q31.3(77,100,000–85,800,000), ×2 hmz | 8.7 | Additional findings | VUS | ||||||
| ‘069 | amniotic fluid | consanguineous marriage | 9p22.3p13.1(16,315,832–38,771,831)×2 hmz | 22.4 | 9p22.3p13.1(14,800,000–39,200,000), ×2 hmz | 24.4 | Consistently | VUS | |
| 9q21.11q21.13(71,100,000–78,500,000), ×2 hmz | 7.4 | Additional findings | VUS | ||||||
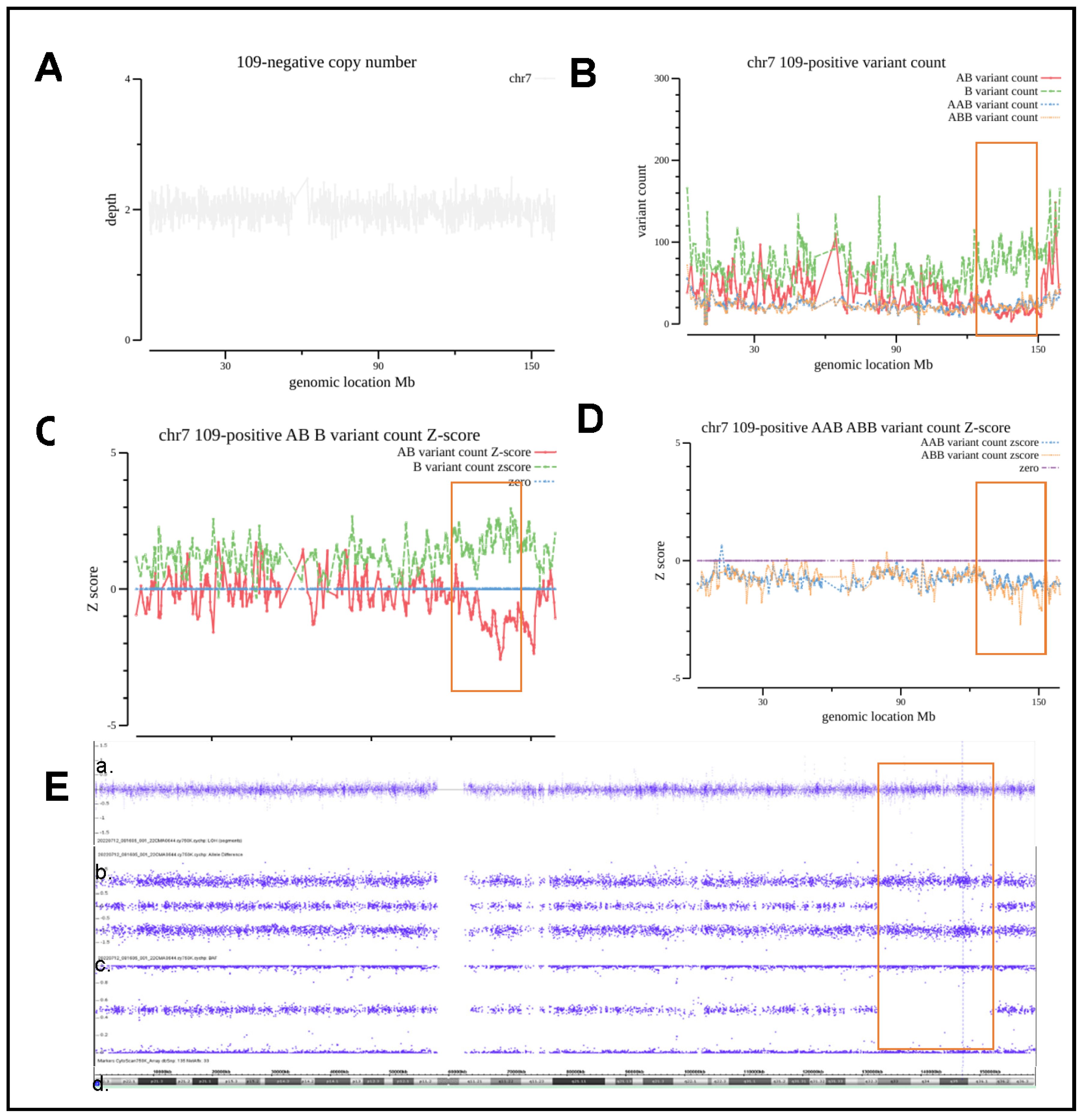
| ‘109 | amniotic fluid | advanced maternal age | 7q32.3q36.1(132,165,146–151,810,715)×2 hmz | 19.6 | 7q32.3q34(132,300,000–142,000,000), ×2 hmz | 9.7 | Consistently | VUS | Silver–Russell syndrome |
| 7q34q36.1(143,100,000–151,500,000), ×2 hmz | 8.4 | Consistently | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lü, Y.; Jiang, Y.; Zhou, X.; Hao, N.; Lü, G.; Guo, X.; Guo, R.; Liu, W.; Xu, C.; Chang, J.; et al. Evaluation and Analysis of Absence of Homozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis. Diagnostics 2023, 13, 560. https://doi.org/10.3390/diagnostics13030560
Lü Y, Jiang Y, Zhou X, Hao N, Lü G, Guo X, Guo R, Liu W, Xu C, Chang J, et al. Evaluation and Analysis of Absence of Homozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis. Diagnostics. 2023; 13(3):560. https://doi.org/10.3390/diagnostics13030560
Chicago/Turabian StyleLü, Yan, Yulin Jiang, Xiya Zhou, Na Hao, Guizhen Lü, Xiangxue Guo, Ruidong Guo, Wenjie Liu, Chenlu Xu, Jiazhen Chang, and et al. 2023. "Evaluation and Analysis of Absence of Homozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis" Diagnostics 13, no. 3: 560. https://doi.org/10.3390/diagnostics13030560
APA StyleLü, Y., Jiang, Y., Zhou, X., Hao, N., Lü, G., Guo, X., Guo, R., Liu, W., Xu, C., Chang, J., Li, M., Zhang, H., Zhou, J., Zhang, W., & Qi, Q. (2023). Evaluation and Analysis of Absence of Homozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis. Diagnostics, 13(3), 560. https://doi.org/10.3390/diagnostics13030560