
Illuminating the Genetic Basis of Congenital Heart Disease in Patients with Kabuki Syndrome

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Data Collection
Molecular Analysis
2.2. Literature Review
2.3. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuroki, Y.; Suzuki, Y.; Chyo, H.; Hata, A.; Matsui, I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 1981, 99, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Niikawa, N.; Matsuura, N.; Fukushima, Y.; Ohsawa, T.; Kajii, T. Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981, 99, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Banka, S.; Bjornsson, H.T.; Bodamer, O.; Chudley, A.E.; Harris, J.; Kawame, H.; Lanpher, B.C.; Lindsley, A.W.; Merla, G.; et al. Kabuki syndrome: International consensus diagnostic criteria. J. Med. Genet. 2019, 56, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Dentici, M.L.; Di Pede, A.; Lepri, F.R.; Gnazzo, M.; Lombardi, M.H.; Auriti, C.; Petrocchi, S.; Pisaneschi, E.; Bellacchio, E.; Capolino, R.; et al. Kabuki syndrome: Clinical and molecular diagnosis in the first year of life. Arch. Dis. Child. 2015, 100, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, H.; Su, C.; Ouyang, Q.; Cao, B.; Wang, J.; Gong, C.; Chen, R. Kabuki syndrome: Novel pathogenic variants, new phenotypes and review of literature. Orphanet J. Rare Dis. 2019, 14, 255. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Marino, B.; Toscano, A.; Giannotti, A.; Dallapiccola, B. Congenital heart defects in Kabuki syndrome. Am. J. Med. Genet. 2001, 100, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Abd El Moneim, A.; Aleck, K.; Aughton, D.J.; Baumann, C.; Braddock, S.R.; Gillessen-Kaesbach, G.; Graham, J.M.; Grebe, T.A.; Gripp, K.W.; et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am. J. Med. Genet. A 2005, 132A, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Schrander-Stumpel, C.T.; Spruyt, L.; Curfs, L.M.; Defloor, T.; Schrander, J.J. Kabuki syndrome: Clinical data in 20 patients, literature review, and further guidelines for preventive management. Am. J. Med. Genet. A 2005, 132A, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Niikawa, N.; Kuroki, Y.; Kajii, T.; Matsuura, N.; Ishikiriyama, S.; Tonoki, H.; Ishikawa, N.; Yamada, Y.; Fujita, M.; Umemoto, H. Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62 patients. Am. J. Med. Genet. 1988, 31, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Philip, N.; Meinecke, P.; David, A.; Dean, J.; Ayme, S.; Clark, R.; Gross-Kieselstein, E.; Hosenfeld, D.; Moncla, A.; Muller, D. Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 16 non-Japanese cases. Clin. Dysmorphol. 1992, 1, 63–77. [Google Scholar] [CrossRef]
- Galán-Gómez, E.; Cardesa-García, J.J.; Campo-Sampedro, F.M.; Salamanca-Maesso, C.; Martínez-Frías, M.L.; Frías, J.L. Kabuki make-up (Niikawa-Kuroki) syndrome in five Spanish children. Am. J. Med. Genet. 1995, 59, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.N. Thirteen cases of Niikawa-Kuroki syndrome: Report and review with emphasis on medical complications and preventive management. Am. J. Med. Genet. 1998, 79, 112–120. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, M.C.; Buckingham, K.J.; Ng, S.B.; Ming, J.E.; Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Bigham, A.W.; Tabor, H.K.; Mefford, H.C.; et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A 2011, 155A, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bögershausen, N.; Alanay, Y.; Simsek Kiper, P.O.; Plume, N.; Keupp, K.; Pohl, E.; Pawlik, B.; Rachwalski, M.; Milz, E.; et al. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011, 130, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Micale, L.; Augello, B.; Fusco, C.; Selicorni, A.; Loviglio, M.N.; Silengo, M.C.; Reymond, A.; Gumiero, B.; Zucchetti, F.; D’Addetta, E.V.; et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, A.D.; Stegmann, A.P.; Blok, M.J.; Tserpelis, D.; Posma-Velter, C.; Detisch, Y.; Smeets, E.E.; Wagemans, A.; Schrander, J.J.; van den Boogaard, M.J.; et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011, 32, E2018–E2025. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; van Bon, B.W.; Steehouwer, M.; Rodríguez-Santiago, B.; Simpson, M.; Dias, P.; Anderlid, B.M.; Arts, P.; Bhat, M.; Augello, B.; et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: A genotype-phenotype study. Clin. Genet. 2013, 84, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef]
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013, 34, 108–110. [Google Scholar] [CrossRef]
- Banka, S.; Lederer, D.; Benoit, V.; Jenkins, E.; Howard, E.; Bunstone, S.; Kerr, B.; McKee, S.; Lloyd, I.C.; Shears, D.; et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin. Genet. 2015, 87, 252–258. [Google Scholar] [CrossRef]
- Bögershausen, N.; Tsai, I.C.; Pohl, E.; Kiper, P.Ö.; Beleggia, F.; Percin, E.F.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y.; et al. RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J. Clin. Investig. 2015, 125, 3585–3599. [Google Scholar] [CrossRef]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef]
- Au, P.Y.B.; You, J.; Caluseriu, O.; Schwartzentruber, J.; Majewski, J.; Bernier, F.P.; Ferguson, M.; Care for Rare Canada Consortium; Valle, D.; Parboosingh, J.S.; et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum. Mutat. 2015, 36, 1009–1014. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Digilio, M.C.; Gnazzo, M.; Lepri, F.; Dentici, M.L.; Pisaneschi, E.; Baban, A.; Passarelli, C.; Capolino, R.; Angioni, A.; Novelli, A.; et al. Congenital heart defects in molecularly proven Kabuki syndrome patients. Am. J. Med. Genet. A 2017, 173, 2912–2922. [Google Scholar] [CrossRef]
- Shone, J.D.; Sellers, R.D.; Anderson, R.C.; Adams, P., Jr.; Lillehei, C.W.; Edwards, J.E. The developmental complex of “parachute mitral valve,” supravalvular ring of left atrium, subaortic stenosis, and coarctation of aorta. Am. J. Cardiol. 1963, 11, 714–725. [Google Scholar] [CrossRef]
- Yoon, J.K.; Ahn, K.J.; Kwon, B.S.; Kim, G.B.; Bae, E.J.; Noh, C.I.; Ko, J.M. The strong association of left-side heart anomalies with Kabuki syndrome. Korean J. Pediatr. 2015, 58, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Shahdadpuri, R.; Lynch, S.A.; Murchan, H.; McMahon, C.J. A novel constellation of cardiac findings for Kabuki syndrome: Hypoplastic left heart syndrome and partial anomalous pulmonary venous drainage. Pediatr. Cardiol. 2008, 29, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Moral, S.; Zuccarino, F.; Loma-Osorio, P. Double aortic arch: An unreported anomaly with Kabuki syndrome. Pediatr. Cardiol. 2009, 30, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.M. Congenital heart defects in Kabuki syndrome. Cardiol. J. 2013, 20, 121–124. [Google Scholar] [CrossRef]
- Belengeanu, V.; Rozsnyai, K.; Farcaş, S.; Velea, I.; Fryns, J.P. Familial transmission of a dysmorphic syndrome: A variant example of Kabuki syndrome? Genet. Couns. 2005, 16, 167–171. [Google Scholar] [PubMed]
- McGaughran, J.; Aftimos, S.; Jefferies, C.; Winship, I. Clinical phenotypes of nine cases of Kabuki syndrome from New Zealand. Clin. Dysmorphol. 2001, 10, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Cheon, C.K.; Ko, J.M. Kabuki syndrome: Clinical and molecular characteristics. Korean J. Pediatr. 2015, 58, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.Ö.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation update for Kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked Kabuki syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.; Shears, D.; Benoit, V.; Verellen-Dumoulin, C.; Maystadt, I. A three generation x-linked family with Kabuki syndrome phenotype and a frameshift mutation in KDM6A. Am. J. Med. Genet. A 2014, 164A, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Hancarova, M.; Puchmajerova, A.; Drabova, J.; Karaskova, E.; Vlckova, M.; Sedlacek, Z. Deletions of 9q21.3 including NTRK2 are associated with severe phenotype. Am. J. Med. Genet. A 2015, 167A, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef]
- Ang, S.Y.; Uebersohn, A.; Spencer, C.I.; Huang, Y.; Lee, J.E.; Ge, K.; Bruneau, B.G. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development 2016, 143, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.L.A.; Demarest, B.L.; Tone-Pah-Hote, T.; Tristani-Firouzi, M.; Yost, H.J. Inhibition of Notch signaling rescues cardiovascular development in Kabuki Syndrome. PLoS Biol. 2019, 17, e3000087. [Google Scholar] [CrossRef] [PubMed]
- de la Pompa, J.L.; Epstein, J.A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev. Cell 2012, 22, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Starmer, J.; Yee, D.; Pohlers, M.; Magnuson, T. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014, 10, e1004507. [Google Scholar] [CrossRef]
- Cocciadiferro, D.; Augello, B.; De Nittis, P.; Zhang, J.; Mandriani, B.; Malerba, N.; Squeo, G.M.; Piccinni, B.; Verri, T.; Gelb, B.D.; et al. Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum. Mol. Genet. 2018, 27, 3651–3668. [Google Scholar] [CrossRef] [PubMed]
- Hasami, N.A.; Ko, K.; Kempers, M.J.E.; van Kimmenade, R.R.J.; Geuzebroek, G.S.C. Type A aortic dissection in a 24-year-old patient with Kabuki syndrome. JACC. Case Rep. 2023, 29, 102149. [Google Scholar] [CrossRef]
- Kung, G.C.; Chang, P.M.; Sklansky, M.S.; Randolph, L.M. Hypoplastic left heart syndrome in patients with Kabuki syndrome. Pediatr. Cardiol. 2010, 31, 138–141. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
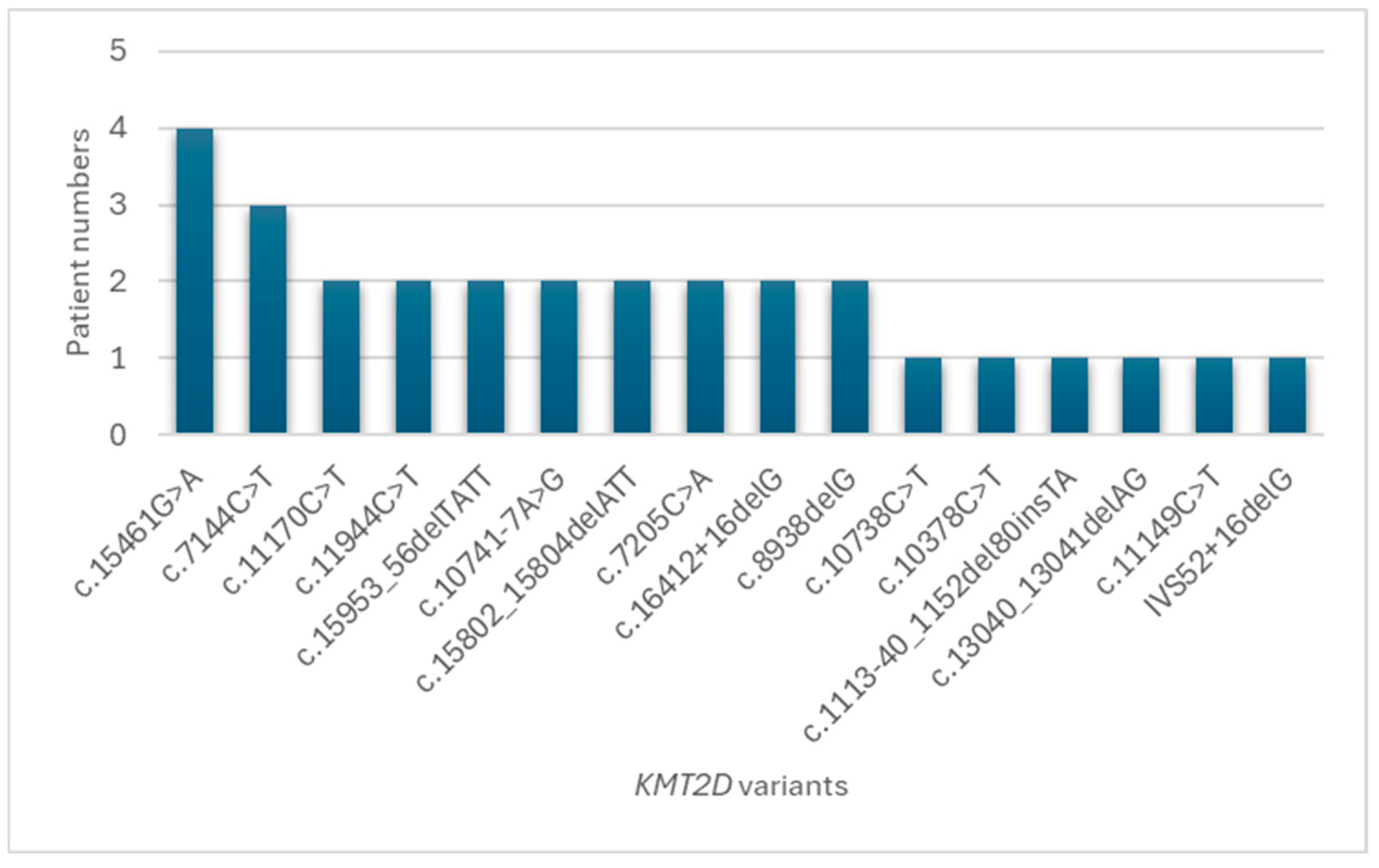
| Patient | Sex | KMT2D cDNA Change | KMT2D Amino Acid Change | ACMG Classification | Congenital Heart Defect |
|---|---|---|---|---|---|
| 1 | M | c.15461G>A | p.Arg5154Gln | Likely Pathogenic | MS, PLSVC, MVP, MR, BAV, CoA, ARSA |
| 2 | M | c.10738C>T | p.Gln3580* | Pathogenic | ASD |
| 3 | M | c.10378C>T | p.Gln3460* | Pathogenic | VSD |
| 4 | M | c.11944C>T | p.Arg3982* | Pathogenic | VSD, redundant TV |
| 5 | M | c.1113-40_1152del80insTA | - | Likely Pathogenic | Vascular ring, PDA |
| 6 | F | c.15953_56delTATT | p.Leu5318Serfs*14 | Pathogenic | ASD |
| 7 | F | c.13040_13041delAG, | p.Gln4347Argfs*24, | Pathogenic, | ASD |
| c.7144C>T | p.Pro2382Ser | Uncertain Significance | |||
| 8 | M | c.10741-7A>G | - | Likely Pathogenic, | PDA |
| c.15802_15804delATT | p.Ile5268del | Pathogenic | |||
| 9 | F | c.7205C>A | p.Ser2402* | Pathogenic | IAA, VSD, PLSVC, SAR, MVP, MR |
| 10 | F | c.15802_15804delATT | p.Ile5268del | Pathogenic | Normal |
| 11 | M | c.7205C>A | p.Ser2402* | Pathogenic | BAV |
| 12 | F | c.15953_56delTATT | p.Leu5318Serfs*14 | Pathogenic | Normal |
| 13 | M | c.15461G>A | p.Arg5154Gln | Likely Pathogenic | Normal |
| 14 | M | c.11149C>T | p.Gln3717* | Likely Pathogenic | Normal |
| 15 | F | c.7144C>T | p.Pro2382Ser | Uncertain Significance | CoA, PDA, LIG, aortic dissection |
| 16 | F | c.7144C>T, c.16412 + 16delG | p.Pro2382Ser, - | Uncertain Significance, Likely Pathogenic | CoA |
| 17 | F | c.15461G>A | p.Arg5154Gln | Likely Pathogenic | Normal |
| 18 | M | c.8938delG | p.Ala2980Profs*24 | Pathogenic | BAV |
| 19 | M | c.11170C>T, c.16412 + 16delG | p.Pro2382Ser, - | Uncertain Significance, Likely Pathogenic | redundant MV, redundant TV, ASD, thick IVS |
| 20 | F | c.8938delG | p.Ala2980Profs*24 | Pathogenic | BAV |
| 21 | F | c.15461G>A | p.Arg5154Gln | Likely Pathogenic | Normal |
| 22 | F | IVS52 + 16delG | - | Likely Pathogenic | ASD |
| 23 | M | c.10741-7A>G | - | Likely Pathogenic | Normal |
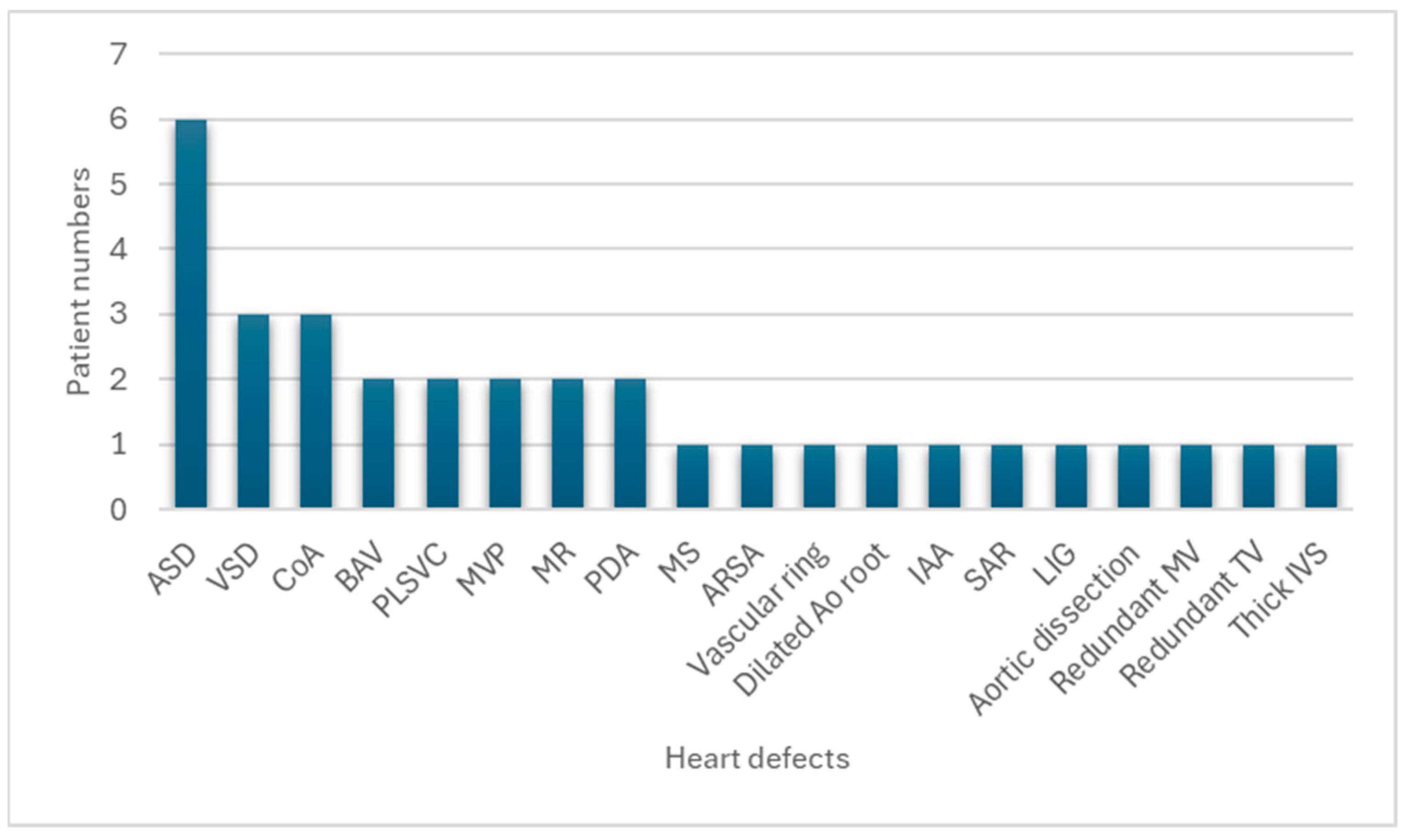
| Congenital Heart Defect | Present Series (Number of Patients with Congenital Heart Defect) | Present Series (%) |
|---|---|---|
| ASD, ostium secundum | 6/16 | 37.5 |
| VSD, perimembranous subaortic | 3/16 | 18.8 |
| Aortic coarctation | 3/16 | 18.8 |
| Bicuspid aortic valve | 2/16 | 12.5 |
| Persistent left superior vena cava | 2/16 | 12.5 |
| Mitral valve prolapse | 2/16 | 12.5 |
| Mitral regurgitation | 2/16 | 12.5 |
| Patent ductus arteriosus | 2/16 | 12.5 |
| Mitral stenosis | 1/16 | 6.3 |
| Aberrant right subclavian artery | 1/16 | 6.3 |
| Vascular ring | 1/16 | 6.3 |
| Dilated aortic root | 1/16 | 6.3 |
| Interrupted aortic arch | 1/16 | 6.3 |
| Subaortic ridge | 1/16 | 6.3 |
| Left isomerism of the heart | 1/16 | 6.3 |
| Aortic dissection | 1/16 | 6.3 |
| Redundant mitral valve | 1/16 | 6.3 |
| Redundant tricuspid valve | 1/16 | 6.3 |
| Thick interventricular septum | 1/16 | 6.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-L.; Chuang, C.-K.; Chen, M.-R.; Lin, J.-L.; Chiu, H.-C.; Chang, Y.-H.; Tu, Y.-R.; Lo, Y.-T.; Lin, H.-Y.; Lin, S.-P. Illuminating the Genetic Basis of Congenital Heart Disease in Patients with Kabuki Syndrome. Diagnostics 2024, 14, 846. https://doi.org/10.3390/diagnostics14080846
Lee C-L, Chuang C-K, Chen M-R, Lin J-L, Chiu H-C, Chang Y-H, Tu Y-R, Lo Y-T, Lin H-Y, Lin S-P. Illuminating the Genetic Basis of Congenital Heart Disease in Patients with Kabuki Syndrome. Diagnostics. 2024; 14(8):846. https://doi.org/10.3390/diagnostics14080846
Chicago/Turabian StyleLee, Chung-Lin, Chih-Kuang Chuang, Ming-Ren Chen, Ju-Li Lin, Huei-Ching Chiu, Ya-Hui Chang, Yuan-Rong Tu, Yun-Ting Lo, Hsiang-Yu Lin, and Shuan-Pei Lin. 2024. "Illuminating the Genetic Basis of Congenital Heart Disease in Patients with Kabuki Syndrome" Diagnostics 14, no. 8: 846. https://doi.org/10.3390/diagnostics14080846
APA StyleLee, C. -L., Chuang, C. -K., Chen, M. -R., Lin, J. -L., Chiu, H. -C., Chang, Y. -H., Tu, Y. -R., Lo, Y. -T., Lin, H. -Y., & Lin, S. -P. (2024). Illuminating the Genetic Basis of Congenital Heart Disease in Patients with Kabuki Syndrome. Diagnostics, 14(8), 846. https://doi.org/10.3390/diagnostics14080846