Circulating Tumor Cell Analysis in Preclinical Mouse Models of Metastasis

Abstract
:1. Introduction
2. The Metastatic Process
3. Clinical Imaging Techniques for Identifying and Tracking Metastasis
3.1. Whole Body-Bone Scanning
3.2. Single Photon Emission Computed Tomography
3.3. Magnetic Resonance Imaging
3.4. Positron Emission Tomography and Computed Tomography
4. Circulating Tumor Cell Analysis Approaches
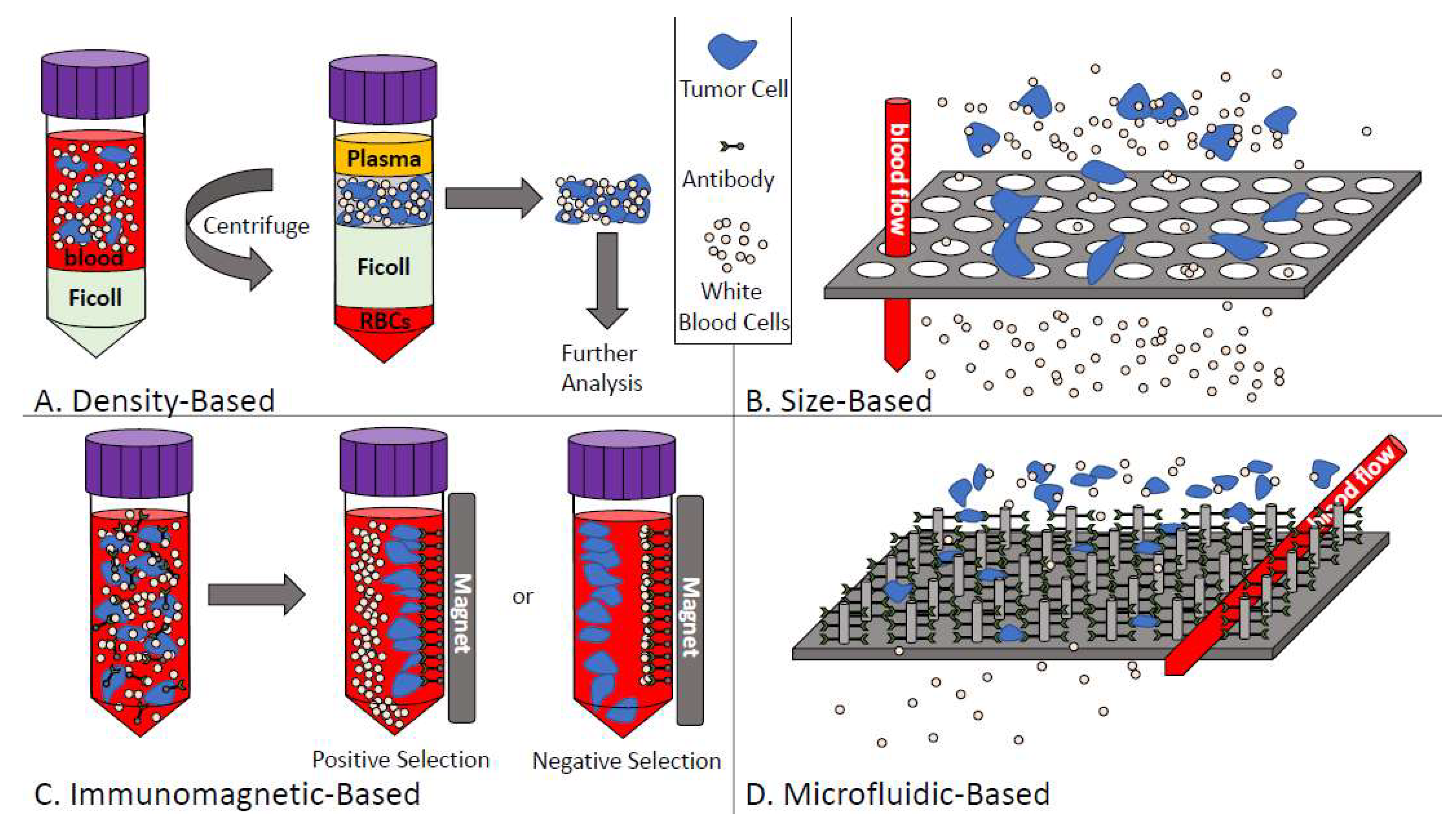
4.1. CTC Enrichment Techniques
4.1.1. Density-Based Enrichment
4.1.2. Size-Based Enrichment
4.1.3. Immunomagnetic-Based Enrichment
4.1.4. Microfluidic-Based Enrichment
4.2. CTC Detection/Characterization Techniques
4.2.1. Protein-Based Detection and Characterization
4.2.2. Nucleic Acid-Based Detection and Characterization
4.3. Additional CTC Analysis Approaches
4.3.1. Dielectrophoresis
4.3.2. Direct Cellular Imaging
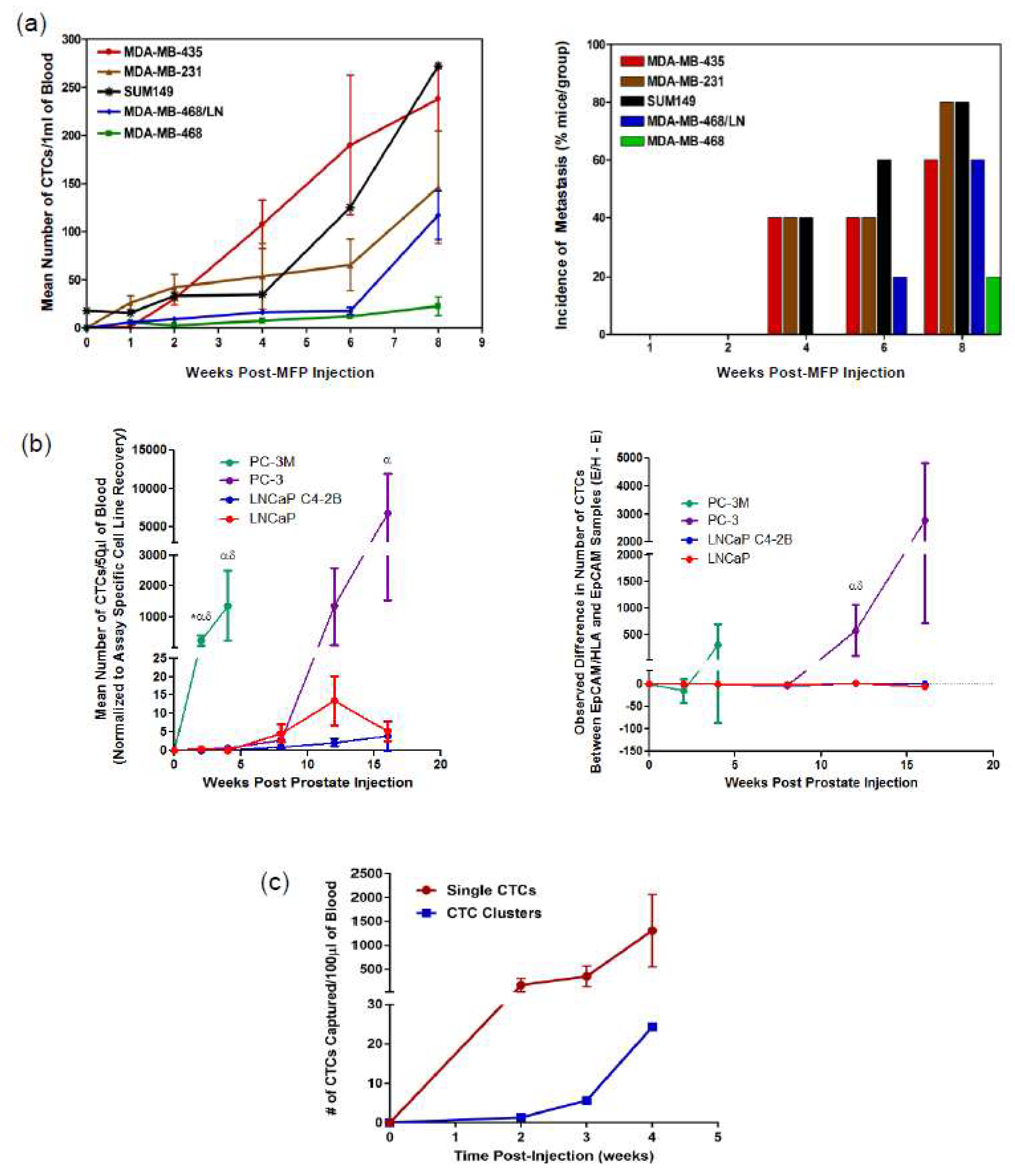
5. Preclinical Models of Metastasis and CTC Generation
5.1. Spontaneous Metastasis Models
5.2. Experimental Metastasis Models
5.3. Genetically Engineered Mouse Metastasis Models
5.4. Patient-Derived Xenograft Models of Metastasis
6. Tracking Metastasis and CTCs in Preclinical Models
6.1. Technologies for Preclinical Evaluation of CTCs
6.1.1. Flow Cytometry
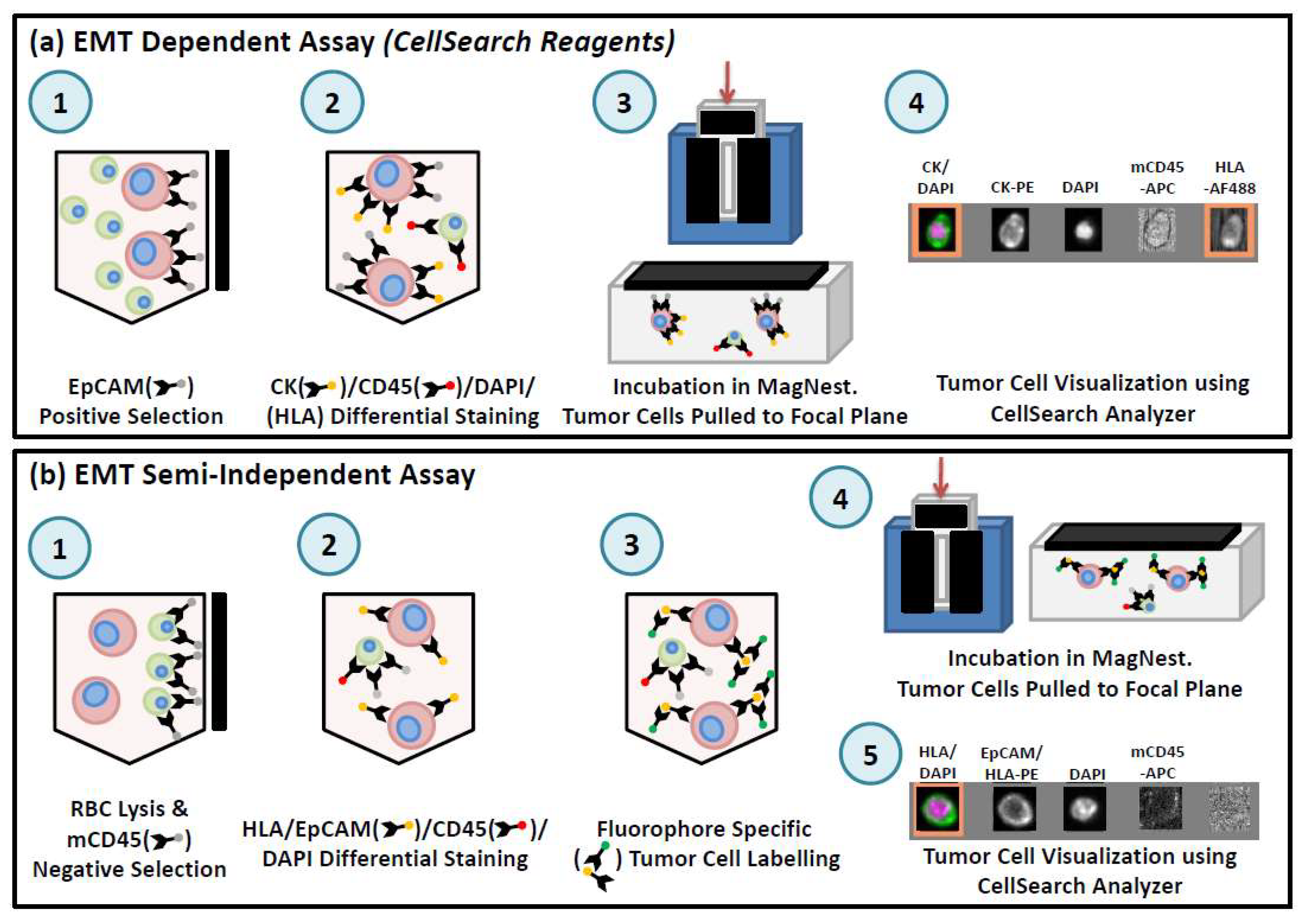
6.1.2. CellSearch®
6.1.3. Parsortix™
6.2. Elucidating the Biology of CTCs and Metastasis via Preclinical Models
7. Conclusions and Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute Cancer Statistics. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 7 February 2018).
- American Cancer Society Survival Rates for Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 7 February 2018).
- American Cancer Society Breast Cancer Survival Rates. Available online: https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html (accessed on 7 February 2018).
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.H.; Petersen, H.; Høilund-Carlsen, P.F.; Jakobsen, J.S.; Gerke, O.; Karstoft, J.; Steffansen, S.I.; Walter, S. Spine metastases in prostate cancer: Comparison of technetium-99m-MDP whole-body bone scintigraphy, [18F]choline positron emission tomography(PET)/computed tomography (CT) and [18F]NaF PET/CT. BJU Int. 2014, 114, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Carloni, V. The metastatic process: Methodological advances and pharmacological challenges. Curr. Med. Chem. 2009, 16, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, R. Patterns of tumor metastasis: Organ selectivity in the spread of cancer cells. Lab. Invest. 1988, 58, 361–364. [Google Scholar] [PubMed]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. Rev. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Albanese, C.; Rodriguez, O.C.; VanMeter, J.; Fricke, S.T.; Rood, B.R.; Lee, Y.; Wang, S.S.; Madhavan, S.; Gusev, Y.; Petricoin, E.F.; et al. Preclinical magnetic resonance imaging and systems biology in cancer research: Current applications and challenges. Am. J. Pathol. 2013, 182, 312–318. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Facts and Figures. 2017. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html (accessed on 7 February 2018).
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Patanaphan, V.; Salazar, O.M.; Risco, R. Breast cancer: Metastatic patterns and their prognosis. South Med. J. 1988, 81, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Schöpfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- The Mayo Clinic. Prostate cancer metastasis: Where does prostate cancer spread? Available online: https://www.mayoclinic.org/diseases-conditions/prostate-cancer/expert-answers/prostate-cancer-metastasis/faq-20058270 (accessed on 28 March 2018).
- National Cancer Institute, Metastatic Cancer. Available online: https://www.cancer.gov/types/metastatic-cancer (accessed on 28 March 2018).
- Doheny, K. The Truth about Whole-Body Scans. Available online: https://www.webmd.com/a-to-z-guides/features/truth-about-whole-body-scans#1 (accessed on 28 March 2018).
- Lin, C.; Luciani, A.; Itti, E.; Haioun, C.; Rahmouni, A. Whole body MRI and PET/CT in haematological malignancies. Cancer Imaging 2007, 7, S88–S93. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014, 74, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Even-Sapir, E.; Metser, U.; Mishani, E.; Lievshitz, G.; Lerman, H.; Leibovitch, I. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP Planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F-fluoride PET, and 18F-fluoride PET/CT. J. Nucl. Med. 2006, 47, 287–297. [Google Scholar] [PubMed]
- Shen, G.; Deng, H.; Hu, S.; Jia, Z. Comparison of choline-PET/CT, MRI, SPECT, and bone scintigraphy in the diagnosis of bone metastases in patients with prostate cancer: A meta-analysis. Skeletal Radiol. 2014, 43, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Sanches, P.G.; Peters, S.; Rossin, R.; Kaijzel, E.L.; Que, I.; Löwik, C.W.G.M.; Grüll, H. Bone metastasis imaging with SPECT/CT/MRI: A preclinical toolbox for therapy studies. Bone 2015, 75, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Ueda, C.E.; Ichiki, W.A.; Ruiz, M.F.; Santos, A.O.; Etchebehere, E.C. 99mTc-DISIDA uptake in liver lesion and pulmonary metastases shown on SPECT/CT in a patient with hepatocellular carcinoma. Clin. Nucl. Med. 2014, 39, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.C.; Xu, Z.; Guley, K.; Yuan, H.; Huang, L. Lipid-calcium phosphate nanoparticles for delivery to the lymphatic system and SPECT/CT imaging of lymph node metastases. Biomaterials 2014, 35, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Mathematics and Physics of Emerging Biomedical Imaging; National Academies Press: Washington, DC, USA, 1996; ISBN 0309552923. [Google Scholar]
- Giovanella, L.; Castellani, M.; Suriano, S.; Ruberto, T.; Ceriani, L.; Tagliabue, L.; Lucignani, G. Multi-field-of-view SPECT is superior to whole-body scanning for assessing metastatic bone disease in patients with prostate cancer. Tumori 2011, 97, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Radiologic Society of North America Magnetic Resonance Imaging (MRI)—Prostate. Available online: https://www.radiologyinfo.org/en/info.cfm?pg=mr_prostate (accessed on 13 February 2018).
- Vargas, H.A.; Schor-Bardach, R.; Long, N.; Kirzner, A.N.; Cunningham, J.D.; Goldman, D.A.; Moskowitz, C.S.; Sosa, R.E.; Sala, E.; Panicek, D.M.; et al. Prostate cancer bone metastases on staging prostate MRI: prevalence and clinical features associated with their diagnosis. Abdom. Radiol. 2017, 42, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Choyke, P.; Coffey, D.S. Challenges in clinical prostate cancer: Role of imaging. Am. J. Roentgenol. 2009, 192, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Larroza, A.; Moratal, D.; Paredes-S?nchez, A.; Soria-Olivas, E.; Chust, M.L.; Arribas, L.A.; Arana, E. Support vector machine classification of brain metastasis and radiation necrosis based on texture analysis in MRI. J. Magn. Reson. Imaging 2015, 42, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Namasivayam, S.; Martin, D.R.; Saini, S. Imaging of liver metastases: MRI. Cancer Imaging 2007, 7, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Society Magnetic Resonance Imaging (MRI). Available online: http://www.cancer.ca/en/cancer-information/diagnosis-and-treatment/tests-and-procedures/magnetic-resonance-imaging-mri/?region=on (accessed on 13 February 2018).
- Radiologic Society of North America Positron Emission Tomography—Computed Tomography (PET/CT). Available online: https://www.radiologyinfo.org/en/info.cfm?pg=pet (accessed on 28 February 2018).
- Rowe, S.P.; Macura, K.J.; Mena, E.; Blackford, A.L.; Nadal, R.; Antonarakis, E.S.; Eisenberger, M.; Carducci, M.; Fan, H.; Dannals, R.F.; et al. PSMA-Based [18F]DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Mol. Imaging Biol. 2016, 18, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Schöder, H.; Larson, S.M. Positron emission tomography for prostate, bladder, and renal cancer. Semin. Nucl. Med. 2004, 34, 274–292. [Google Scholar] [CrossRef] [PubMed]
- Even-Sapir, E.; Metser, U.; Flusser, G.; Zuriel, L.; Kollender, Y.; Lerman, H.; Lievshitz, G.; Ron, I.; Mishani, E. Assessment of malignant skeletal disease: Initial experience with 18F-fluoride PET/CT and comparison between 18F-fluoride PET and 18F-fluoride PET/CT. J. Nucl. Med. 2004, 45, 272–278. [Google Scholar] [PubMed]
- Choi, E.K.; Oh, J.K.; Chung, Y.A. Herpes Zoster Mimicking Breast Cancer With Axillary Lymph Node Metastasis on PET/CT. Clin. Nucl. Med. 2015, 40, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Harsha, D.S.; Moleyar, V.; Bhat, A.; Konda, I.; Bali, A.; Ummer, A. Pattern of Lung Cancer Metastasis Based on PET CT. J. Evid. Based Med. Healthc. 2017, 4, 3108–3111. [Google Scholar]
- Li, M.; Lu, H.; Gao, Y. FDG-anorectic parathyroid carcinoma with FDG-avid bone metastasis on PET/CT images. Clin. Nucl. Med. 2013, 38, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Liu, J.; Wu, C.; Song, S.; Tong, L.; Huang, G.; Feng, Y.; Jiang, Y.; Liu, Y.; Yin, T.; et al. Prognostic significance of (18)FDG PET/CT in colorectal cancer patients with liver metastases: A meta-analysis. Cancer Imaging 2015, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.B.; Aboagye, E.O.; Adams, J.E.; Aerts, H.J.W.L.; Barrington, S.F.; Beer, A.J.; Boellaard, R.; Bohndiek, S.E.; Brady, M.; Brown, G.; et al. Imaging biomarker roadmap for cancer studies. Nat. Rev. Clin. Oncol. 2017, 14, 169. [Google Scholar] [CrossRef] [PubMed]
- Auguste, P.; Barton, P.; Hyde, C.; Roberts, T.E. An economic evaluation of positron emission tomography (PET) and positron emission tomography/computed tomography (PET/CT) for the diagnosis of breast cancer recurrence. Health Technol. Assess. 2011, 15, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Goodale, D.; Xia, Y.; Postenka, C.; Piaseczny, M.M.; Paczkowski, F.; Allan, A.L. Epithelial-to-mesenchymal transition leads to disease-stage differences in circulating tumor cell detection and metastasis in pre-clinical models of prostate cancer. Oncotarget 2016, 7, 76125–76139. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.P.H.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Andree, K.C.; van Dalum, G.; Terstappen, L.W.M.M. Challenges in circulating tumor cell detection by the CellSearch system. Mol. Oncol. 2016, 10, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Gao, P.; Song, Y.; Sun, J.; Chen, X.; Zhao, J.; Xu, H.; Wang, Z. Meta-analysis of the prognostic value of circulating tumor cells detected with the CellSearch System in colorectal cancer. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Benali-Furet, N.; Uzan, G.; Znaty, A.; Ye, Z.; Paolillo, C.; Wang, C.; Austin, L.; Rossi, G.; Fortina, P.; et al. Detection and characterization of circulating tumor associated cells in metastatic breast cancer. Int. J. Mol. Sci. 2016, 17, 1665. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, J.; Kim, S.T.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K. Circulating tumor cells are predictive of poor response to chemotherapy in metastatic gastric cancer. Int. J. Biol. Markers 2015, 30, e382–e386. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.-Y.; Lin, K.-X.; Zhang, Z.; Zhang, D.-J.; Shi, C.-H.; Xiong, M.; Qu, X.-H.; Zhao, X.-H. Monitoring disease progression and treatment efficacy with circulating tumor cells in esophageal squamous cell carcinoma: A case report. World J. Gastroenterol. 2015, 21, 7921–7928. [Google Scholar] [CrossRef] [PubMed]
- Bredemeier, M.; Edimiris, P.; Mach, P.; Kubista, M.; Sjöback, R.; Rohlova, E.; Kolostova, K.; Hauch, S.; Aktas, B.; Tewes, M.; et al. Gene expression signatures in circulating tumor cells correlate with response to therapy in metastatic breast cancer. Clin. Chem. 2017, 63, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Görner, K.; Bachmann, J.; Holzhauer, C.; Kirchner, R.; Raba, K.; Fischer, J.C.; Martignoni, M.E.; Schiemann, M.; Alunni-Fabbroni, M. Genetic analysis of circulating tumor cells in pancreatic cancer patients: A pilot study. Genomics 2015, 106, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed]
- Alunni-Fabbroni, M.; Sandri, M.T. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods 2010, 50, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Allan, A.L. Recent advances in the molecular characterization of circulating tumor cells. Cancers 2014, 6, 595–624. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E.S.; Markou, A. Circulating tumor cells in breast cancer: Detection systems, molecular characterization, and future challenges. Clin. Chem. 2011, 57, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Peeters, D.J.E.; De Laere, B.; Van den Eynden, G.G.; Van Laere, S.J.; Rothé, F.; Ignatiadis, M.; Sieuwerts, A.M.; Lambrechts, D.; Rutten, A.; Van Dam, P.A. Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br. J. Cancer 2013, 108, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abonnenc, M.; Manaresi, N.; Borgatti, M.; Medoro, G.; Fabbri, E.; Romani, A.; Altomare, L.; Tartagni, M.; Rizzo, R.; Baricordi, O.; et al. Programmable interactions of functionalized single bioparticles in a dielectrophoresis-based microarray chip. Anal. Chem. 2013, 85, 8219–8224. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.L.; Graf, R.P.; Landers, M.; Valenta, D.T.; Schroeder, M.; Greene, S.B.; Bales, N.; Dittamore, R.; Marrinucci, D. Analytical Validation and Capabilities of the Epic CTC Platform: Enrichment-Free Circulating Tumour Cell Detection and Characterization. J. Circ. Biomark. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Marrinucci, D.; Bethel, K.; Kolatkar, A.; Luttgen, M.S.; Malchiodi, M.; Baehring, F.; Voigt, K.; Lazar, D.; Nieva, J.; Bazhenova, L.; et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 2012, 9, 016003. [Google Scholar] [CrossRef] [PubMed]
- Goodale, D.; Phay, C.; Postenka, C.O.; Keeney, M.; Allan, A.L. Characterization of tumor cell dissemination patterns in preclinical models of cancer metastasis using flow cytometry and laser scanning cytometry. Cytometry A 2009, 75A, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.C.; Yan, J.; Liu, G.D.; Tan, X.Y.; Weng, X.F.; Wu, W.Z.; Zhou, J.; Wei, X. Bin Real-time monitoring of rare circulating hepatocellular carcinoma cells in an orthotopic model by in vivo flow cytometry assesses resection on metastasis. Cancer Res. 2012, 72, 2683–2691. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R. Technical considerations for studying cancer metastasis in vivo. Clin. Exp. Metastasis 1997, 15, 272–306. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.; Herrera, S.; Christiny, P.; Shaw, C.; Creighton, C.J.; Mitchell, T.; Bhat, R.; Zhang, X.; Mao, S.; Dobrolecki, L.E.; et al. Circulating and disseminated tumor cells from breast cancer patient-derived xenograft-bearing mice as a novel model to study metastasis. Breast Cancer Res. 2015, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, C.A.; Liu, S.Z.; Wilkerson, C.L.; Ramani, V.C.; Barzanian, N.A.; Huang, K.W.; Che, J.; Chiu, M.W.; Vuppalapaty, M.; Dimmick, A.M.; et al. Fast and label-free isolation of circulating tumor cells from blood: from a research microfluidic platform to an automated fluidic instrument, VTX-1 liquid biopsy system. SLAS Technol. 2018, 23, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Kinders, R.J.; Kummar, S.; Gupta, V.; Hasegawa, D.; Menachery, A.; Lawrence, S.M.; Wang, L.; Ferry-Galow, K.; Davis, D.; et al. Antibody-independent capture of circulating tumor cells of non-epithelial origin with the ApoStream® system. PLoS ONE 2017, 12, e0175414. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Goodale, D.; Keeney, M.; Allan, A.L. Image Cytometry Analysis of Circulating Tumor Cells; Academic Press: Cambridge, MA, USA, 2011; Volume 102, ISBN 9780123749123. [Google Scholar]
- Allan, A.L.; Vantyghem, S.A; Tuck, A.B.; Chambers, A.F.; Chin-Yee, I.H.; Keeney, M. Detection and quantification of circulating tumor cells in mouse models of human breast cancer using immunomagnetic enrichment and multiparameter flow cytometry. Cytometry A 2005, 65, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Hedley, B.D.; Keeney, M.; Allan, A.L. Adaptation of semiautomated circulating tumor cell (CTC) assays for clinical and preclinical research applications. J. Vis. Exp. 2014, 84, e51248. [Google Scholar] [CrossRef] [PubMed]
- Chudziak, J.; Burt, D.J.; Mohan, S.; Rothwell, D.G.; Mesquita, B.; Antonello, J.; Dalby, S.; Ayub, M.; Priest, L.; Carter, L.; et al. Clinical evaluation of a novel microfluidic device for epitope-independent enrichment of circulating tumour cells in patients with small cell lung cancer. Analyst 2016, 141, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Hvichia, G.E.; Parveen, Z.; Wagner, C.; Janning, M.; Quidde, J.; Stein, A.; Müller, V.; Loges, S.; Neves, R.P.L.; Stoecklein, N.H.; et al. A novel microfluidic platform for size and deformability based separation and the subsequent molecular characterization of viable circulating tumor cells. Int. J. Cancer 2016, 138, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Parsortix Test Procedure. Available online: https://angleplc.com/pasortix-technology/test-procedure/ (accessed on 23 April 2018).
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mao, X.; Imrali, A.; Syed, F.; Mutsvangwa, K.; Berney, D.; Cathcart, P.; Hines, J.; Shamash, J.; Lu, Y.-J. Optimization and Evaluation of a Novel Size Based Circulating Tumor Cell Isolation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Peddibhotla, S.; Yin, W.; Scamardo, A.T.; George, G.C.; Hong, D.S.; Marchetti, D. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Pantel, K. Tumor Cell Dissemination: Emerging Biological Insights from Animal Models and Cancer Patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Approach | Sample Volume Requirement * | Ability for CTC Quantification | High Specificity | High Sensitivity | Labor-Intensive/Challenging | Down-Stream Analysis |
|---|---|---|---|---|---|---|---|
| Protein-Based | Immunofluorescence | Small | Yes | Yes | No | Yes | Yes |
| Flow Cytometry | Medium | Yes | Yes | No | Yes | Yes | |
| CellSearch® | Large | Yes | Yes | Yes | No | No | |
| CTC-Chip/iChip | Large | Yes | Yes | Yes | Yes | Yes | |
| AdnaTest | Large | No | Yes | Yes | No | No | |
| Isoflux | Large | Yes | Yes | Yes | No | Yes | |
| Nucleic Acid-Based | RT-PCR | Small | No | No | Yes | No | No |
| qRT-PCR | Small | No | No | Yes | No | No | |
| Next-Gen Sequencing | Small | No | Yes | Yes | Yes | Yes |
| Feature | Flow Cytometry | CellSearch® | Parsortix™ |
|---|---|---|---|
| Sensitivity | ☑ | ☑☑☑ | ☑☑☑ |
| Captures cells independent of phenotype | ☒ | ☒ | ☑ |
| Ability for downstream analysis | ☑ | ☒ | ☑☑☑ |
| Accurate low blood volume analysis | ☒ | ☑ | ☑☑☑ |
| Ease of process | ☒ | ☒ | ☑ |
| Low cost | ☑☑☑ | ☒ | ☑☑ |
| Support for research/flexibility | ☑☑ | ☒ | ☑☑☑ |
| Clinical relevance | ☒ | ☑☑ (FDA) | ☑ (CE Mark) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitz, J.; Lowes, L.E.; Goodale, D.; Allan, A.L. Circulating Tumor Cell Analysis in Preclinical Mouse Models of Metastasis. Diagnostics 2018, 8, 30. https://doi.org/10.3390/diagnostics8020030
Kitz J, Lowes LE, Goodale D, Allan AL. Circulating Tumor Cell Analysis in Preclinical Mouse Models of Metastasis. Diagnostics. 2018; 8(2):30. https://doi.org/10.3390/diagnostics8020030
Chicago/Turabian StyleKitz, Jenna, Lori E. Lowes, David Goodale, and Alison L. Allan. 2018. "Circulating Tumor Cell Analysis in Preclinical Mouse Models of Metastasis" Diagnostics 8, no. 2: 30. https://doi.org/10.3390/diagnostics8020030
APA StyleKitz, J., Lowes, L. E., Goodale, D., & Allan, A. L. (2018). Circulating Tumor Cell Analysis in Preclinical Mouse Models of Metastasis. Diagnostics, 8(2), 30. https://doi.org/10.3390/diagnostics8020030