Identification and Characterization of BTD Gene Mutations in Jordanian Children with Biotinidase Deficiency

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Sample Collection
2.3. DNA Extraction and Genotyping
2.4. Enzyme Assay
3. Results
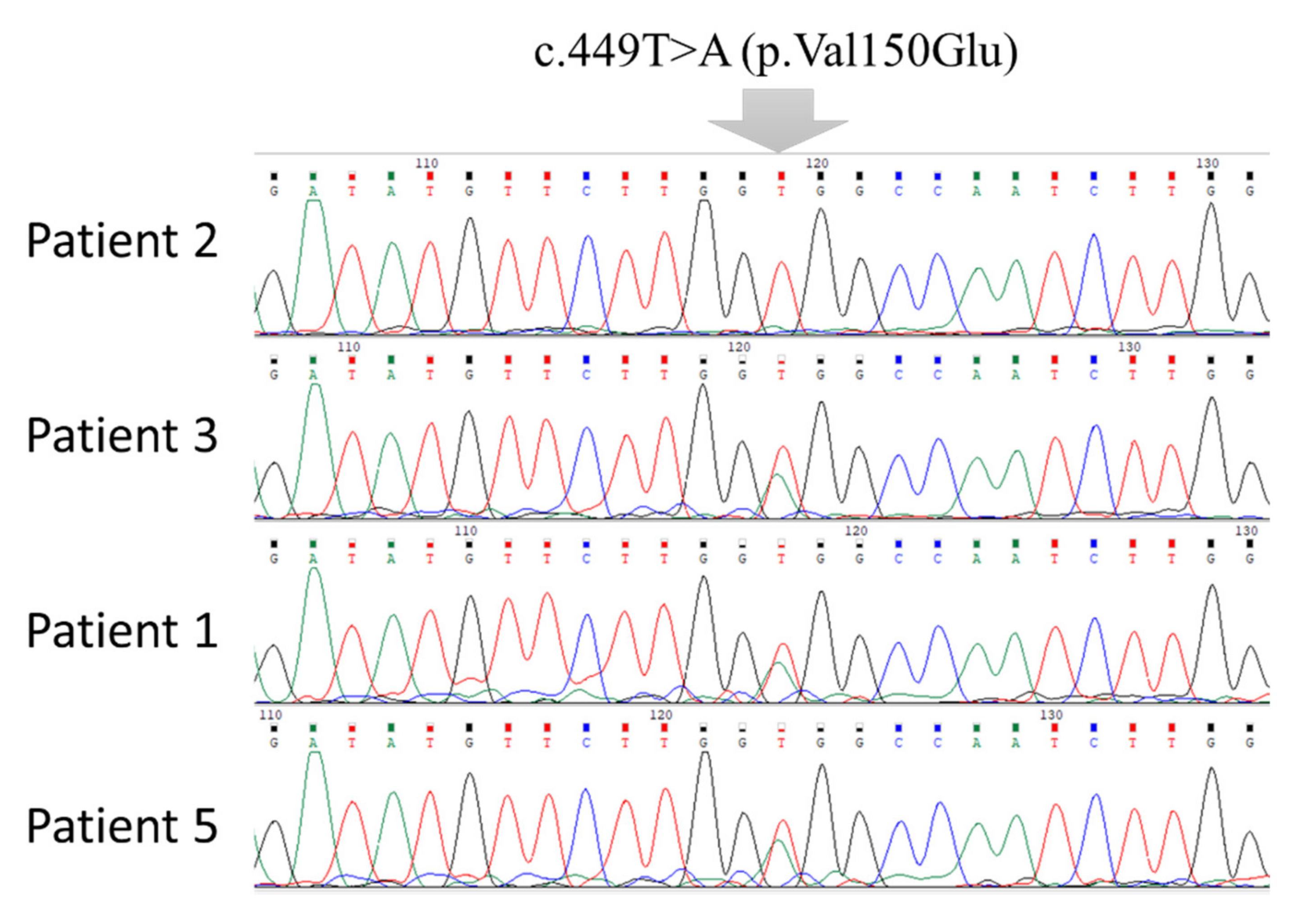
3.1. Clinical Characterization Data of BTD Patients and Mutation Analysis
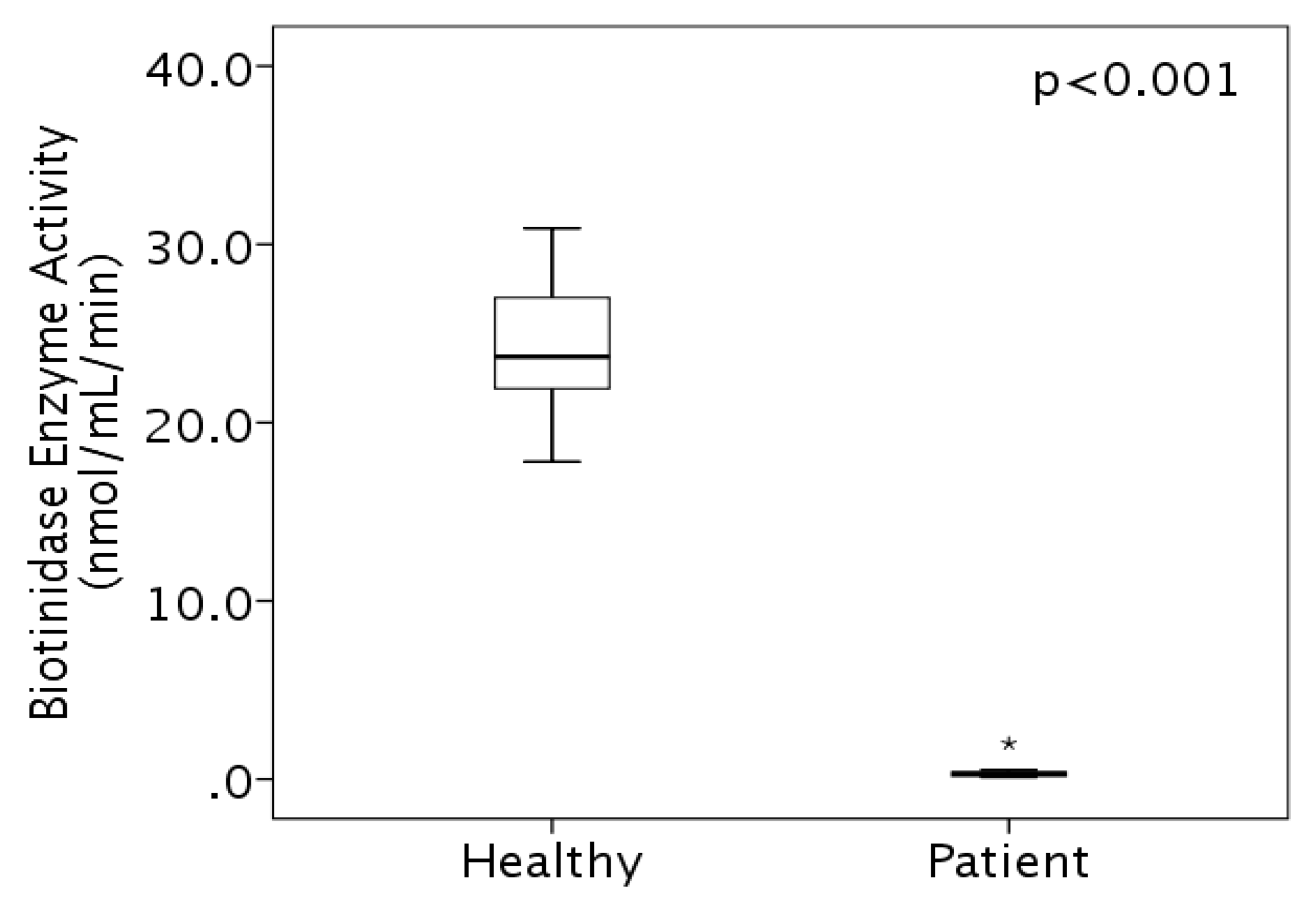
3.2. Enzyme Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tiar, A.; Mekki, A.; Nagara, M.; Rhouma, F.B.; Messaoud, O.; Halim, N.B.; Kefi, R.; Hamlaoui, M.T.; Lebied, A.; Abdelhak, S. Biotinidase deficiency: Novel mutations in Algerian patients. Gene 2014, 536, 193–196. [Google Scholar] [CrossRef]
- Thodi, G.; Schulpis, K.H.; Molou, E.; Georgiou, V.; Loukas, Y.L.; Dotsikas, Y.; Papadopoulos, K.; Biti, S. High incidence of partial biotinidase deficiency cases in newborns of Greek origin. Gene 2013, 524, 361–362. [Google Scholar] [CrossRef]
- Karaca, M.; Özgül, R.K.; Ünal, Ö.; Yücel-Yılmaz, D.; Kılıç, M.; Hişmi, B.; Tokatlı, A.; Coşkun, T.; Dursun, A.; Sivri, H.S. Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening. Eur. J. Pediatr. 2015, 174, 1077–1084. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; Solórzano-Vargas, R.S.; del Río, A.L. Biotin in metabolism and its relationship to human disease. Arch. Med. Res. 2002, 33, 439–447. [Google Scholar] [CrossRef]
- Küry, S.; Ramaekers, V.; Bézieau, S.; Wolf, B. Clinical utility gene card for: Biotinidase deficiency. Eur. J. Hum. Genet. 2012, 20, 592. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Spencer, L.; Nahhas, F.; Miller, J.; Fribley, A.; Feldman, G.; Conway, R.; Wolf, B. Novel mutations causing biotinidase deficiency in individuals identified by newborn screening in Michigan including an unique intronic mutation that alters mRNA expression of the biotinidase gene. Mol. Genet. Metab. 2014, 112, 242–246. [Google Scholar] [CrossRef]
- Mikati, M.A.; Zalloua, P.; Karam, P.; Habbal, M.Z.; Rahi, A.C. Novel Mutation Causing Partial Biotinidase Deficiency in a Syrian Boy With Infantile Spasms and Retardation. We report a case of partial biotinidase deficiency. J. Child Neurol. 2006, 21, 978–981. [Google Scholar] [CrossRef]
- U.S. National Library of Midecine, Genetics Home Reference, Health Conditions, Biotinidase Deficiency. Available online: https://ghr.nlm.nih.gov/condition/biotinidase-deficiency (accessed on 9 September 2019).
- Cowan, T.M.; Kazerouni, N.N.; Dharajiya, N.; Lorey, F.; Roberson, M.; Hodgkinson, C.; Schrijver, I. Increased incidence of profound biotinidase deficiency among Hispanic newborns in California. Mol. Genet. Metab. 2012, 106, 485–487. [Google Scholar] [CrossRef]
- Wolf, B. The neurology of biotinidase deficiency. Mol. Genet. Metab. 2011, 104, 27–34. [Google Scholar] [CrossRef]
- Heard, G.S.; Secor McVoy, J.R.; Wolf, B. A screening method for biotinidase deficiency in newborns. Clin. Chem. 1984, 30, 125–127. [Google Scholar] [CrossRef]
- Wolf, B. Biotinidase deficiency: If you have to have an inherited metabolic disease, this is the one to have. Genet. Med. 2012, 14, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Hsu, R.H.; Chien, Y.H.; Hwu, W.L.; Chang, I.F.; Ho, H.C.; Chou, S.P.; Huang, T.M.; Lee, N.C. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J. Rare Dis. 2019, 14, 6. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Ahmadabadi, F.; Jafari, N.; Jabbehdari, S.; Alaee, M.R.; Ghofrani, M.; Taghdiri, M.M.; Tonekaboni, S.H. Biotinidase deficiency: A reversible neurometabolic disorder (An Iranian pediatric case series). Iran. J. Child Neurol. 2013, 7, 47–52. [Google Scholar]
- Singh, A.; Lomash, A.; Pandey, S.; Kapoor, S. Clinical, biochemical and outcome profile of biotinidase deficient patients from tertiary centre in Northern India. J. Clin. Diagn. Res. 2015, 9, SC08–SC10. [Google Scholar] [CrossRef]
- Sivri, H.S.K.; Genç, G.A.; Tokatlý, A.; Dursun, A.; Cothkun, T.; Aydýn, H.Ý.; Sennarolu, L.; Belgin, E.; Jensen, K.; Wolf, B. Hearing Loss in Biotinidase Deficiency: Genotype-Phenotype Correlation. J. Pediatr. 2007, 150, 439–442. [Google Scholar] [CrossRef]
- Kasapkara, Ç.S.; Akar, M.; Özbek, M.N.; Tüzün, H.; Aldudak, B.; Baran, R.T.; Tanyalçin, T. Mutations in BTD gene causing biotinidase deficiency: A regional report. J. Pediatr. Endocrinol. Metab. 2015, 28, 421–424. [Google Scholar] [CrossRef]
- Wolf, B.; Jensen, K.; Hüner, G.; Demirkol, M.; Baykal, T.; Divry, P.; Rolland, M.O.; Perez-Cerda, C.; Ugarte, M.; Straussberg, R.; et al. Seventeen novel mutations that cause profound biotinidase deficiency. Mol. Genet. Metab. 2002, 77, 108–111. [Google Scholar] [CrossRef]
- Iqbal, F.; Item, C.B.; Vilaseca, M.A.; Jalan, A.; Mühl, A.; Couce, M.L.; Duat, A.; Delgado, M.P.; Bosch, J.; Puche, A.; et al. The identification of novel mutations in the biotinidase gene using denaturing high pressure liquid chromatography (dHPLC). Mol. Genet. Metab. 2010, 100, 42–45. [Google Scholar] [CrossRef]
- Senanayake, D.N.; Jasinge, E.A.; Pindolia, K.; Wanigasinghe, J.; Monaghan, K.; Suchy, S.F.; Wei, S.; Jaysena, S.; Wolf, B. First contiguous gene deletion causing biotinidase deficiency: The enzyme deficiency in three Sri Lankan children. Mol. Genet. Metab. 2015, 2, 81–84. [Google Scholar] [CrossRef]
- Pomponio, R.J.; Reynolds, T.R.; Cole, H.; Buck, G.A.; Wolf, B. Mutational hotspot in the human biotinidase gene causes profound biotinidase deficiency. Nat. Genet. 1995, 11, 96–98. [Google Scholar] [CrossRef]
- Wolf, B.; Jensen, K.P.; Barshop, B.; Blitzer, M.; Carlson, M.; Goudie, D.R.; Gokcay, G.H.; Demirkol, M.; Baykal, T.; Demir, F.; et al. Biotinidase deficiency: Novel mutations and their biochemical and clinical correlates. Hum. Mutat. 2005, 25, 413. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, X.; Sheng, H.; Cai, Y.; Yin, X.; Chen, X.; Su, L.; Lu, Z.; Zeng, C.; Li, X.; et al. Clinical features, BTD gene mutations, and their functional studies of eight symptomatic patients with biotinidase deficiency from Southern China. Am. J. Med. Genet. Part A 2018, 176, 589–596. [Google Scholar] [CrossRef]
- Asgari, A.; Dehnabeh, S.R.; Zargari, M.; Khani, S.; Mozafari, H.; Varasteh, A.; Keyfi, F.; Barzegari, M.; Hasanzaeh, R.; Khatami, S. Clinical, biochemical and genetic analysis of biotinidase deficiency in Iranian population. Arch. Iran. Med. 2016, 19, 774–778. [Google Scholar]
- Szymańska, E.; Sredzińska, M.; Ługowska, A.; Pajdowska, M.; Rokicki, D.; Tylki-Szymańska, A. Outcomes of oral biotin treatment in patients with biotinidase deficiency—Twenty years follow-up. Mol. Genet. Metab. 2015, 5, 33–35. [Google Scholar] [CrossRef]
- Pomponio, R.J.P.; Oskun, T.C.; Emirkol, M.D.; Okatli, A.T.; Zalp, I.O. Novel mutations cause biotinidase deüciency in Turkish children. J. Inherit. Metab. Dis. 2000, 23, 120–128. [Google Scholar] [CrossRef]
- Baykal, T.; Gokcay, G.; Gokdemir, Y.; Demir, F.; Seckin, Y.; Demirkol, M.; Jensen, K.; Wolf, B. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J. Inherit. Metab. Dis. 2005, 28, 903–912. [Google Scholar] [CrossRef]
- Ohlsson, A.; Guthenberg, C.; Holme, E.; von Döbeln, U. Profound biotinidase deficiency: A rare disease among native Swedes. J. Inherit. Metab. Dis. 2010, 33, 175–180. [Google Scholar] [CrossRef] [Green Version]
- David, J.; Chrastina, P.; Pešková, K.; Kožich, V.; Friedecký, D.; Adam, T.; Hlídková, E.; Vinohradská, H.; Novotná, D.; Hedelová, M.; et al. Epidemiology of rare diseases detected by newborn screening in the Czech Republic. Cent. Eur. J. Public Health 2019, 27, 153–159. [Google Scholar] [CrossRef]
- Carvalho, N.O.; del Castillo, D.M.; Januário, J.N.; Starling, A.L.; Arantes, R.R.; Norton, R.C.; Viana, M.B. Novel mutations causing biotinidase deficiency in individuals identified by the newborn screening program in Minas Gerais, Brazil. Am. J. Med Genet. Part A 2019, 179, 978–982. [Google Scholar] [CrossRef]
- Borsatto, T.; Sperb-Ludwig, F.; Blom, H.J.; Schawartz, I.V.D. Effect of BTD gene variants on in vitro biotinidase activity. Mol. Genet. Metab. 2019, 127, 361–367. [Google Scholar] [CrossRef]
- Borsatto, T.; Sperb-Ludwig, F.; Lima, S.E.; Carvalho, M.R.; Fonseca, P.A.; Camelo, J.S., Jr.; Ribeiro, E.M.; de Medeiros, P.F.; Lourenco, C.M.; de Souza, C.F.; et al. Biotinidase deficiency: Genotype-biochemical phenotype association in Brazilian patients. PLoS ONE 2017, 12, e0177503. [Google Scholar]
- AL-Eitan, L.; Haddad, Y. Emergence of pharmacogenomics in academic medicine and public health in Jordan: History, present state and prospects. Curr. Pharm. Person. Med. 2014, 12, 167–175. [Google Scholar] [CrossRef]
- AL-Eitan, L.; Tarkhan, A. Practical challenges and translational issues in pharmacogenomics and personalized medicine from 2010 onwards. Curr. Pharm. Person. Med. 2014, 14, 7–17. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Exon No. † | Forward Primer | Reverse Primer | Product Size |
|---|---|---|---|
| 1 | AGAATGTAAACACGCGCGTT | AGAGCGTAAACCACAAAGCG | 465 bp |
| 2 | TCTTTGAGCCGCAGTATCAC | TTCAGAGGGTGGTAGGAAGC | 554 bp |
| 3 | ATGAATGCAGCGGTTCTTCC | TGGCACATGGATCTTTGGGA | 360 bp |
| 4a | GGTGGTCTCAATCTCCTGAC | GTGGAGATAGCCTTCCTTTC | 892 bp |
| 4b | GCGATCCGTACTGTGAGAAG | AGACCAATCGCATACTGAGAGA | 818 bp |
| Family No. | Patient No. | Gender † | Age of Diagnosis | Age at Biotin Administration | Consanguinity | Neurological Traits ‡ | Dermatological Traits | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizures | Hypotonia | Ataxia | Speech Delay | Hearing Loss | Mental Retardation | Conjunctivitis | Alopecia | Rash | ||||||
| 1 | 1 | M | 2 weeks | Since birth | Yes | No | No | No | No | No | No | No | No | No |
| 2 | F | 6 months | 6 months | Yes | Yes | No | No | No | No | No | No | No | ||
| 2 | 3 | M | 3 months | 3 months | Yes | Yes | Yes | No | Yes | Yes | No | No | Yes | No |
| 3 | 4 | F | 27 months | 27 months | Yes | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes |
| 4 | 5 | F | 8 months | 9 months | Yes | Yes | Yes | NA | NA | No | No | No | No | No |
| 5 | 6 | F | 21 months | 21 months | Yes | Yes | Yes | Yes | No | No | No | No | Yes | No |
| 6 | 7 | M | 12 months | 4 months | Far relatives | Yes | Yes | No | No | Yes | No | No | No | No |
| 8 | M | 4 months | 3 months | No | No | No | No | No | No | No | No | No | ||
| 9 | M | 9 months | 3.5 months | No | No | No | No | No | No | No | No | No | ||
| 7 | 10 | F | 3 years | 3 years | Yes | Yes | Yes | Yes | Yes | No | No | No | Yes | No |
| Family No. | Patient No. | Exon | Mutation | Nucleotide Change † | Variant Type | Protein Change | Enzyme Assay (nmol/min/mL) |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 3 | rs397514356 | del C hetero | Frameshift | Phe111Leu | 0.2 |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 2 | 3 | rs397514356 | del C hetero | Frameshift | Phe111Leu | 0.3 | |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 2 | 3 | 2 | rs104893687 | C/T | Missense | Arg79Cys | 0.3 |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 4 | rs397514345 | A/G | Missense | Tyr434Cys | |||
| 3 | 4 | 3 | rs397514356 | del C homo | Frameshift | Phe111Leu | 0.5 |
| 4 | 5 | 4 | c.449T>A | T/A | Missense | Val150Glu | 0.17 |
| 4 | rs397514411 | Ins C hetero | Frameshift | Leu402Pro | |||
| 5 | 6 | 3 | rs397514356 | del C homo | Frameshift | Phe111Leu | 0.3 |
| 6 | 7 | 2 | rs80338684 | del/ins homo GCGGCTG/TCC | Frameshift | Cys13Phe | 0.4 |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 8 | 2 | rs80338684 | del/ins hetero GCGGCTG/TCC | Frameshift | Cys13Phe | 0.1 | |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 9 | 2 | rs80338684 | del/ins homo GCGGCTG/TCC | Frameshift | Cys13Phe | 2.0 | |
| 4 | c.449T>A | T/A | Missense | Val150Glu | |||
| 7 | 10 | 4 | rs397514411 | Ins C homo | Frameshift | Leu402Pro | 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL-Eitan, L.N.; Alqa’qa’, K.; Amayreh, W.; Khasawneh, R.; Aljamal, H.; Al-Abed, M.; Haddad, Y.; Rawashdeh, T.; Jaradat, Z.; Haddad, H. Identification and Characterization of BTD Gene Mutations in Jordanian Children with Biotinidase Deficiency. J. Pers. Med. 2020, 10, 4. https://doi.org/10.3390/jpm10010004
AL-Eitan LN, Alqa’qa’ K, Amayreh W, Khasawneh R, Aljamal H, Al-Abed M, Haddad Y, Rawashdeh T, Jaradat Z, Haddad H. Identification and Characterization of BTD Gene Mutations in Jordanian Children with Biotinidase Deficiency. Journal of Personalized Medicine. 2020; 10(1):4. https://doi.org/10.3390/jpm10010004
Chicago/Turabian StyleAL-Eitan, Laith N., Kifah Alqa’qa’, Wajdi Amayreh, Rame Khasawneh, Hanan Aljamal, Mamoon Al-Abed, Yazan Haddad, Tamara Rawashdeh, Zaher Jaradat, and Hazem Haddad. 2020. "Identification and Characterization of BTD Gene Mutations in Jordanian Children with Biotinidase Deficiency" Journal of Personalized Medicine 10, no. 1: 4. https://doi.org/10.3390/jpm10010004
APA StyleAL-Eitan, L. N., Alqa’qa’, K., Amayreh, W., Khasawneh, R., Aljamal, H., Al-Abed, M., Haddad, Y., Rawashdeh, T., Jaradat, Z., & Haddad, H. (2020). Identification and Characterization of BTD Gene Mutations in Jordanian Children with Biotinidase Deficiency. Journal of Personalized Medicine, 10(1), 4. https://doi.org/10.3390/jpm10010004