Biomarkers for Alzheimer’s Disease Early Diagnosis


Abstract
:1. Introduction
1.1. Pathophysiology of AD and Clinical Manifestations
1.2. Diagnostic Tools
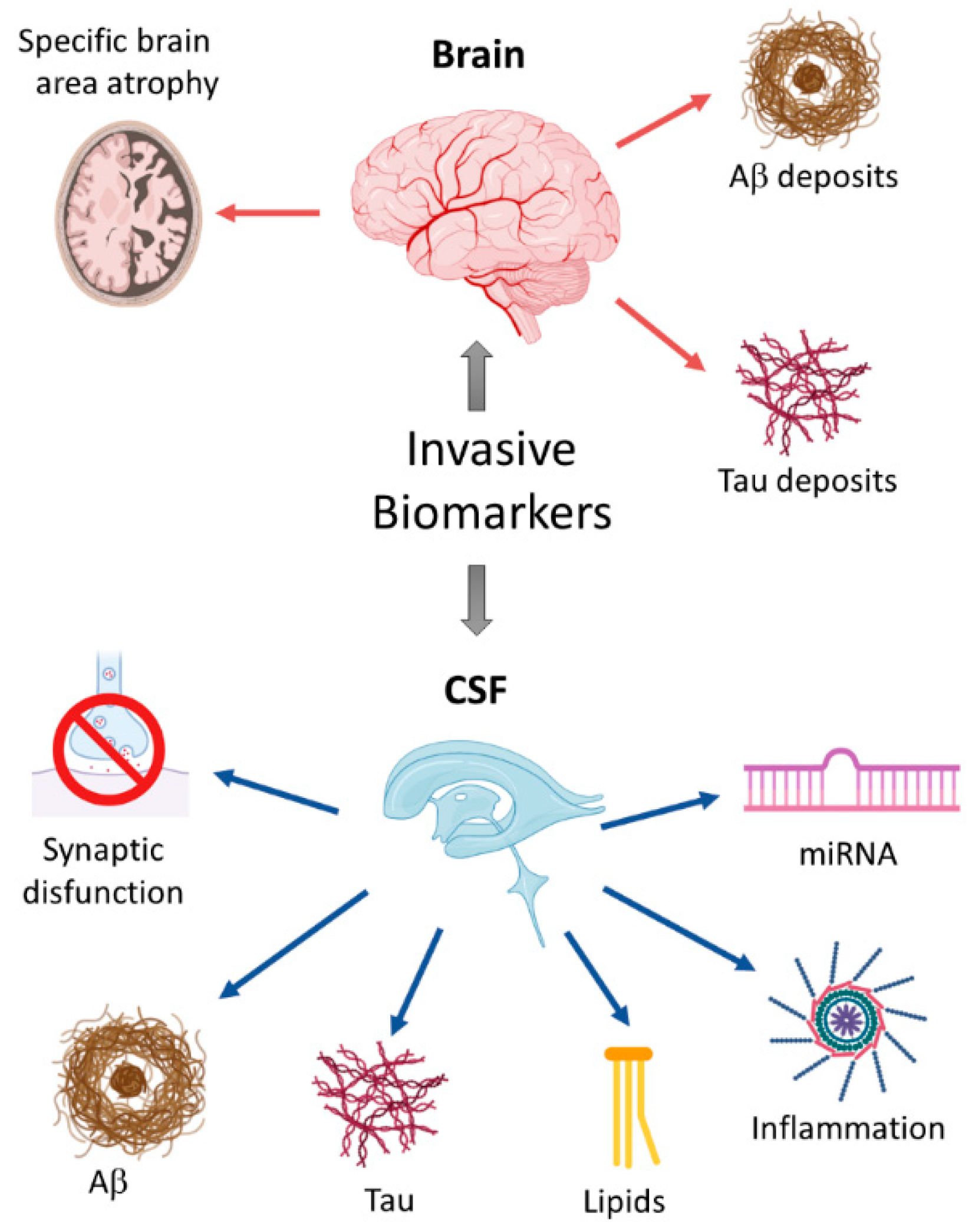
2. Invasive Biomarkers
2.1. Changes in Specific Brain Areas as Early Biomarkers
2.2. Cerebrospinal Fluid
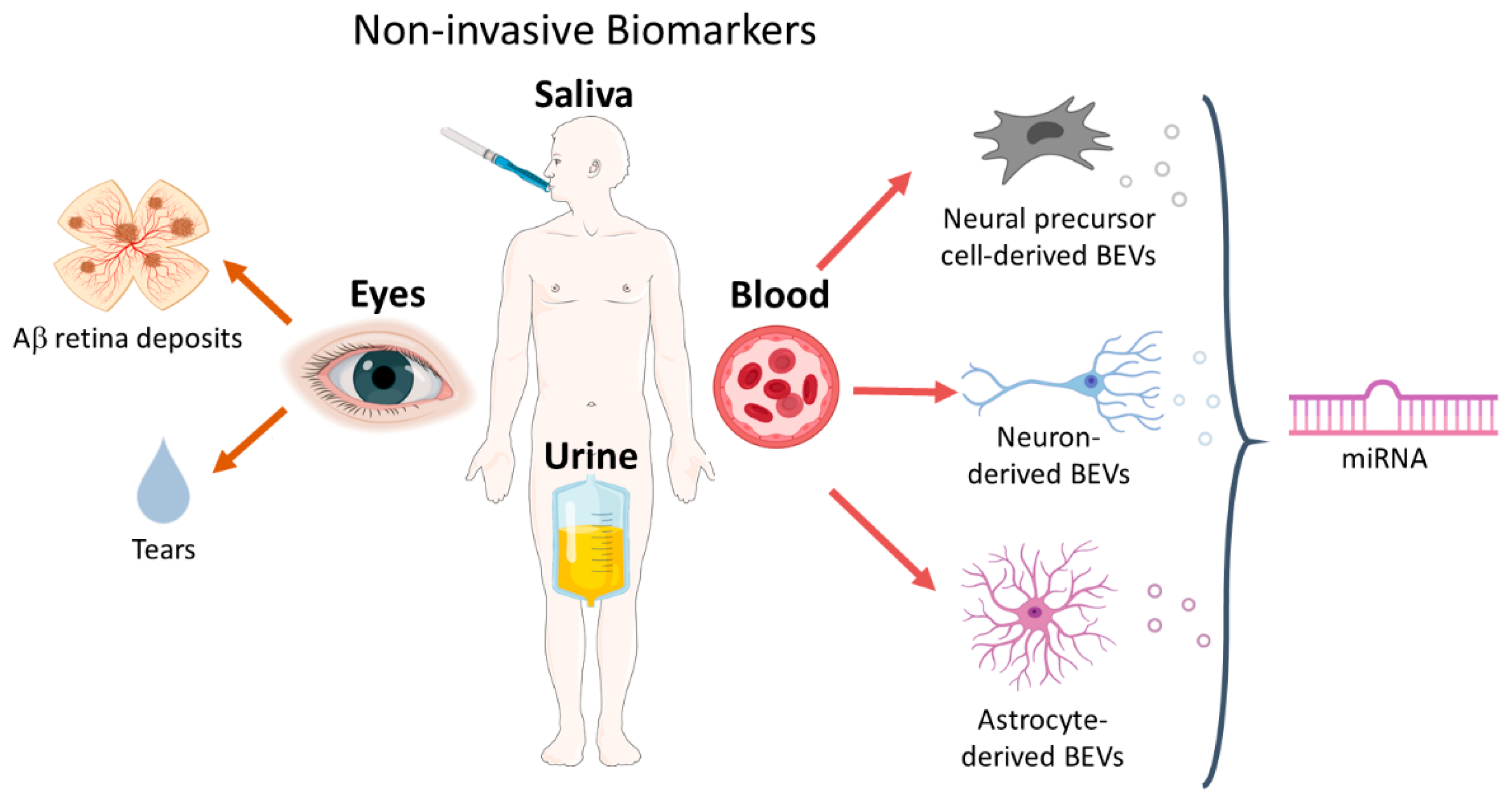
3. Noninvasive Biomarkers
3.1. Blood Biomarkers
3.1.1. Neuron-Derived BEVs in Blood
3.1.2. Neural Precursor Cell-Derived BEVs in Blood
3.1.3. Astrocyte-Derived BEVs in Blood
3.1.4. MicroRNA Cargo of Blood-Isolated EVs
3.2. Ocular Biomarkers
3.3. Salivary Biomarkers
3.4. Urine Biomarkers
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dubois, B.; Feldman, H.H.; Jacova, C.; DeKosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Mueller, S.G.; Schuff, N.; Yaffe, K.; Madison, C.; Miller, B.; Weiner, M.W. Hippocampal atrophy patterns in mild cognitive impairment and alzheimer’s disease. Hum. Brain Mapp. 2010, 31, 1339–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Joie, R.; Perrotin, A.; De La Sayette, V.; Egret, S.; Doeuvre, L.; Belliard, S.; Eustache, F.; Desgranges, B.; Chételat, G. Hippocampal subfield volumetry in mild cognitive impairment, Alzheimer’s disease and semantic dementia. NeuroImage Clin. 2013, 3, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Pluta, J.; Yushkevich, P.; Das, S.; Wolk, D. In vivo Analysis of Hippocampal Subfield Atrophy in Mild Cognitive Impairment via Semi-Automatic Segmentation of T2-Weighted MRI. J. Alzheimer’s Dis. 2012, 31, 85–99. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Morawski, M.; Jäger, C.; Gertz, H.J. Early neurone loss in Alzheimer’s disease: Cortical or subcortical? Acta Neuropathol. Commun. 2015, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K. Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr. Opin. Neurol. 2012, 25, 708–714. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Fernández-Dueñas, V.; Carmona, M.; Escobar, L.A.; Torrejón-Escribano, B.; Aso, E.; Ciruela, F.; Ferrer, I. Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol. Appl. Neurobiol. 2017, 43, 373–392. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Ehrenberg, A.J.; Nguy, A.K.; Theofilas, P.; Dunlop, S.; Suemoto, C.K.; Di Lorenzo Alho, A.T.; Leite, R.P.; Diehl Rodriguez, R.; Mejia, M.B.; Rüb, U.; et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 393–408. [Google Scholar] [CrossRef]
- Stratmann, K.; Heinsen, H.; Korf, H.-W.; Del Turco, D.; Ghebremedhin, E.; Seidel, K.; Bouzrou, M.; Grinberg, L.T.; Bohl, J.; Wharton, S.B.; et al. Precortical Phase of Alzheimer’s Disease (AD)-Related Tau Cytoskeletal Pathology. Brain Pathol. 2016, 26, 371–386. [Google Scholar] [CrossRef]
- Theofilas, P.; Ehrenberg, A.J.; Dunlop, S.; Di Lorenzo Alho, A.T.; Nguy, A.; Leite, R.E.P.; Rodriguez, R.D.; Mejia, M.B.; Suemoto, C.K.; Ferretti-Rebustini, R.E.D.L.; et al. Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimer’s Dement. 2017, 13, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyness, S.A.; Zarow, C.; Chui, H.C. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: A meta-analysis. Neurobiol. Aging 2003, 24, 1–23. [Google Scholar] [CrossRef]
- Betts, M.J.; Cardenas-Blanco, A.; Kanowski, M.; Jessen, F.; Düzel, E. In vivo MRI assessment of the human locus coeruleus along its rostrocaudal extent in young and older adults. Neuroimage 2017, 163, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Priovoulos, N.; Jacobs, H.I.L.; Ivanov, D.; Uludağ, K.; Verhey, F.R.J.; Poser, B.A. High-resolution in vivo imaging of human locus coeruleus by magnetization transfer MRI at 3T and 7T. Neuroimage 2018, 168, 427–436. [Google Scholar] [CrossRef]
- Tondelli, M.; Wilcock, G.K.; Nichelli, P.; de Jager, C.A.; Jenkinson, M.; Zamboni, G. Structural MRI changes detectable up to ten years before clinical Alzheimer’s disease. Neurobiol. Aging 2012, 33, 825.e25–825.e36. [Google Scholar] [CrossRef]
- Bernard, C.; Helmer, C.; Dilharreguy, B.; Amieva, H.; Auriacombe, S.; Dartigues, J.F.; Allard, M.; Catheline, G. Time course of brain volume changes in the preclinical phase of Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 143–151.e1. [Google Scholar] [CrossRef]
- Smith, C.D.; Chebrolu, H.; Wekstein, D.R.; Schmitt, F.A.; Jicha, G.A.; Cooper, G.; Markesbery, W.R. Brain structural alterations before mild cognitive impairment. Neurology 2007, 68, 1268–1273. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Dutton, R.A.; Dinov, I.D.; Hayashi, K.M.; Toga, A.W.; Cummings, J.L.; Thompson, P.M. Conversion of mild cognitive impairment to alzheimer disease predicted by hippocampal atrophy maps. Arch. Neurol. 2006, 63, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Petersen, R.C.; Xu, Y.C.; O’Brien, P.C.; Smith, G.E.; Ivnik, R.J.; Boeve, B.F.; Waring, S.C.; Tangalos, E.G.; Kokmen, E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999, 52, 1397–1403. [Google Scholar] [CrossRef] [Green Version]
- Korf, E.S.C.; Wahlund, L.O.; Visser, P.J.; Scheltens, P. Medial temporal lobe atrophy on MRI predicts dementia in patients with mild cognitive impairment. Neurology 2004, 63, 94–100. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Mosconi, L.; Thompson, P.M.; Green, A.E.; Hwang, K.S.; Ramirez, A.; Mistur, R.; Tsui, W.H.; de Leon, M.J. Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol. Aging 2010, 31, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csernansky, J.G.; Wang, L.; Swank, J.; Miller, J.P.; Gado, M.; McKeel, D.; Miller, M.I.; Morris, J.C. Preclinical detection of Alzheimer’s disease: Hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005, 25, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Reiman, E.M.; Alexander, G.E.; Caselli, R.J.; Gerkin, R.; Bandy, D.; Domb, A.; Osborne, D.; Fox, N.; Crum, W.R.; et al. Correlations between apolipoprotein E ε4 gene dose and whole brain atrophy rates. Am. J. Psychiatry 2007, 164, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Mormino, E.C. APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology 2017, 89, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE Isoforms Differentially Regulate Brain Amyloid- Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [Green Version]
- Reiman, E.M.; Uecker, A.; Caselli, R.J.; Lewis, S.; Bandy, D.; De Leon, M.J.; De Santi, S.; Convit, A.; Osborne, D.; Weaver, A.; et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer’s disease. Ann. Neurol. 1998, 44, 288–291. [Google Scholar] [CrossRef]
- Fang, Y.; Du, N.; Xing, L.; Duo, Y.; Zheng, L. Evaluation of hippocampal volume and serum brain-derived neurotrophic factor as potential diagnostic markers of conversion from amnestic mild cognitive impairment to Alzheimer disease A STROBE-compliant article. Medicine (United States) 2019, 98. [Google Scholar] [CrossRef]
- Finnema, S.J.; Nabulsi, N.B.; Eid, T.; Detyniecki, K.; Lin, S.F.; Chen, M.K.; Dhaher, R.; Matuskey, D.; Baum, E.; Holden, D.; et al. Imaging synaptic density in the living human brain. Sci. Transl. Med. 2016, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Lista, S.; Hampel, H. Synaptic degeneration and neurogranin in the pathophysiology of Alzheimer’s disease. Expert Rev. Neurother. 2017, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Valkimadi, P.E.; Pagano, G.; Cousins, O.; Dervenoulas, G.; Politis, M. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 5424–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain Imaging in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarrisa, A.; Serrano-Aguilar, P. Meta-review of CSF core biomarkers in Alzheimer’s disease: The state-of-the-art after the new revised diagnostic criteria. Front. Aging Neurosci. 2014, 6, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Fishman, R. Cerebrospinal fluid in diseases of the nervous system. WB Saunders Co. 1992, 183–252. [Google Scholar]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; Dekosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Kester, M.I.; Van Der Vlies, A.E.; Blankenstein, M.A.; Pijnenburg, Y.A.L.; Van Elk, E.J.; Scheltens, P.; Van Der Flier, W.M. CSF biomarkers predict rate of cognitive decline in Alzheimer disease. Neurology 2009, 73, 1353–1358. [Google Scholar] [CrossRef]
- Martínez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ayllón, M.-S.; Campanari, M.-L.; Brinkmalm, G.; Rábano, A.; Alom, J.; Saura, C.A.; Andreasen, N.; Blennow, K.; Sáez-Valero, J. CSF Presenilin-1 complexes are increased in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewers, M.; Zhong, Z.; Bürger, K.; Wallin, A.; Blennow, K.; Teipel, S.J.; Shen, Y.; Hampel, H. Increased CSF-BACE 1 activity is associated with ApoE-ε4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain 2008, 131, 1252–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetterberg, H.; Andreasson, U.; Hansson, O.; Wu, G.; Sankaranarayanan, S.; Andersson, M.E.; Buchhave, P.; Londos, E.; Umek, R.M.; Minthon, L.; et al. Elevated cerebrospinal fluid BACE1 activity in incipient alzheimer disease. Arch. Neurol. 2008, 65, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Duits, F.H.; Brinkmalm, G.; Teunissen, C.E.; Brinkmalm, A.; Scheltens, P.; Van Der Flier, W.M.; Zetterberg, H.; Blennow, K. Synaptic proteins in CSF as potential novel biomarkers for prognosis in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Insel, P.; Nosheny, R.; Zetterberg, H.; Trojanowski, J.Q.; Shaw, L.M.; Tosun, D.; Weiner, M. CSF protein biomarkers predicting longitudinal reduction of CSF β-amyloid42 in cognitively healthy elders. Transl. Psychiatry 2013, 3. [Google Scholar] [CrossRef]
- Kester, M.I.; Teunissen, C.E.; Sutphen, C.; Herries, E.M.; Ladenson, J.H.; Xiong, C.; Scheltens, P.; Van Der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimer’s Res. Ther. 2015, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A Novel Prognostic Fluid Biomarker for Preclinical Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Bettcher, B.M.; Johnson, S.C.; Fitch, R.; Casaletto, K.B.; Heffernan, K.S.; Asthana, S.; Zetterberg, H.; Blennow, K.; Carlsson, C.M.; Neuhaus, J.; et al. Cerebrospinal Fluid and Plasma Levels of Inflammation Differentially Relate to CNS Markers of Alzheimer’s Disease Pathology and Neuronal Damage. J. Alzheimer’s Dis. 2018, 62, 385–397. [Google Scholar] [CrossRef]
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal Fluid Biomarkers for Understanding Multiple Aspects of Alzheimer’s Disease Pathogenesis; Springer International Publishing: New York, NY, USA, 2019; Volume 76, ISBN 0001801903040. [Google Scholar]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current State of Alzheimer’s Fluid Biomarkers; Springer: Berlin/Heidelberg, Germany, 2018; Volume 136, ISBN 0040101819. [Google Scholar]
- Zetterberg, H.; Skillbäck, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60. [Google Scholar] [CrossRef]
- Schmidt, F.M.; Mergl, R.; Stach, B.; Jahn, I.; Gertz, H.J.; Schönknecht, P. Elevated levels of cerebrospinal fluid neuron-specific enolase (NSE) in Alzheimer’s disease. Neurosci. Lett. 2014, 570, 81–85. [Google Scholar] [CrossRef]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; Van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer’s disease. J. Neural Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal fluid sphingolipids, β-amyloid, and tau in adults at risk for Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2486–2494. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.T.; Chen-Plotkin, A.; Arnold, S.E.; Grossman, M.; Clark, C.M.; Shaw, L.M.; Pickering, E.; Kuhn, M.; Chen, Y.; Mccluskey, L.; et al. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010, 119, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desikan, R.S.; Thompson, W.K.; Holland, D.; Hess, C.P.; Brewer, J.B.; Zetterberg, H.; Blennow, K.; Andreassen, O.A.; McEvoy, L.K.; Hyman, B.T.; et al. Heart fatty acid binding protein and Aβ-associated Alzheimer’s neurodegeneration. Mol. Neurodegener. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiasserini, D.; Parnetti, L.; Andreasson, U.; Zetterberg, H.; Giannandrea, D.; Calabresi, P.; Blennow, K. CSF levels of heart fatty acid binding protein are altered during early phases of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 22, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provost, P. Interpretation and applicability of microrna datato the context of Alzheimer’s and age-related diseases. Aging (Albany. NY) 2010, 2, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukiw, W.J.; Andreeva, T.V.; Grigorenko, A.P.; Rogaev, E.I. Studying micro RNA function and dysfunction in Alzheimer’s disease. Front. Genet. 2013, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef]
- Cortez, M.A.; Calin, G.A. MicroRNA identification in plasma and serum: A new tool to diagnose and monitor diseases. Expert Opin. Biol. Ther. 2009, 9, 703–711. [Google Scholar] [CrossRef]
- Gallego, J.A.; Gordon, M.L.; Claycomb, K.; Bhatt, M.; Lencz, T.; Malhotra, A.K. In Vivo MicroRNA Detection and Quantitation in Cerebrospinal Fluid. J. Mol. Neurosci. 2012, 47, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Ferrara, N.; Rengo, G. The emerging role of microRNAs in Alzheimer’s disease. Front. Physiol. 2015, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. MicroRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar]
- Lukiw, W.J.; Alexandrov, P.N.; Zhao, Y.; Hill, J.M.; Bhattacharjee, S. Spreading of Alzheimer’s disease inflammatory signaling through soluble micro-RNA. Neuroreport 2012, 23, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavaraju, M.; De Lencastre, A. Alzheimer’s disease: Presence and role of microRNAs. Biomol. Concepts 2016, 7, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: MiR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Jäkel, L.; Bruinsma, I.B.; Claassen, J.A.; Kuiperij, H.B.; Verbeek, M.M. MicroRNA-29a Is a Candidate Biomarker for Alzheimer’s Disease in Cell-Free Cerebrospinal Fluid. Mol. Neurobiol. 2016, 53, 2894–2899. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Tan, E.K.; Zeng, L. microRNAs and Neurodegenerative Diseases. In Advances in Experimental Medicine and Biology; Academic Press: Cambridge, MA, USA, 2015; Chapter 6; pp. 85–105. ISBN 978-3-319-22671-2. [Google Scholar]
- Zhang, F.; Gradinaru, V.; Adamantidis, A.R.; Durand, R.; Airan, R.D.; de Lecea, L.; Deisseroth, K. Optogenetic interrogation of neural circuits: Technology for probing mammalian brain structures. Nat. Protoc. 2010, 5, 439–456. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Nakao, H.; Kiyonari, H.; Abe, T.; Aizawa, S. MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J. Neurosci. 2011, 31, 3407–3422. [Google Scholar] [CrossRef]
- Vassar, R.; Kovacs, D.M.; Yan, R.; Wong, P.C. The β-secretase enzyme BACE in health and Alzheimer’s disease: Regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009, 29, 12787–12794. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, Y.; Zhang, L.; Dong, Y.; Ji, H.; Shen, L. The potential markers of circulating micrornas and long non-coding RNAs in Alzheimer’s disease. Aging Dis. 2019, 10, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Skoog, I.; Gustafson, D. Update on hypertension and Alzheimer’s disease. Neurol. Res. 2006, 28, 605–611. [Google Scholar] [CrossRef]
- Sancesario, G.; Bernardini, S. AD biomarker discovery in CSF and in alternative matrices. Clin. Biochem. 2019, 72, 52–57. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimer’s Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Z.J.; Zhang, Y.; Chen, Y.; Zhao, H.; Stephenson, M.C.; Ho, N.R.Y.; Chen, Y.; Chung, J.; Reilhac, A.; Loh, T.P.; et al. Subtyping of circulating exosome-bound amyloid β reflects brain plaque deposition. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; Van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Xia, W.; Yang, T.; Shankar, G.; Smith, I.M.; Shen, Y.; Walsh, D.M.; Selkoe, D.J. A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer Disease. Arch. Neurol. 2009, 66, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Tatebe, H.; Kasai, T.; Ohmichi, T.; Kishi, Y.; Kakeya, T.; Waragai, M.; Kondo, M.; Allsop, D.; Tokuda, T. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef]
- Hrubešová, K.; Fousková, M.; Habartová, L.; Fišar, Z.; Jirák, R.; Raboch, J.; Setnička, V. Search for biomarkers of Alzheimer‘s disease: Recent insights, current challenges and future prospects. Clin. Biochem. 2019, 72, 39–51. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers with Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Pláteník, J.; Fišar, Z.; Buchal, R.; Jirák, R.; Kitzlerová, E.; Zvěřová, M.; Raboch, J. GSK3β, CREB, and BDNF in peripheral blood of patients with alzheimer’s disease and depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 50, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.B.; Doecke, J.D.; Hone, E.; Pedrini, S.; Laws, S.M.; Thambisetty, M.; Bush, A.I.; Rowe, C.C.; Villemagne, V.L.; Ames, D.; et al. Plasma apolipoprotein J as a potential biomarker for Alzheimer’s disease: Australian Imaging, Biomarkers and Lifestyle study of aging. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongbloed, W.; Van Dijk, K.D.; Mulder, S.D.; Van De Berg, W.D.J.; Blankenstein, M.A.; Van Der Flier, W.; Veerhuis, R. Clusterin Levels in Plasma Predict Cognitive Decline and Progression to Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Yan, Z.; Zhou, Z.; Wu, Q.; Chen, Z.B.; Koo, E.H.; Zhong, S. Presymptomatic Increase of an Extracellular RNA in Blood Plasma Associates with the Development of Alzheimer’s Disease. Curr. Biol. 2020, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2020, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Li, J.; Huang, L.; Chen, X.; Li, D.; Wang, T.; Hu, C.; Xu, J.; Zhang, C.; Zen, K.; et al. Serum MicroRNA profiles serve as novel biomarkers for the diagnosis of alzheimer’s disease. Dis. Markers 2015, 2015. [Google Scholar] [CrossRef]
- Kenny, A.; McArdle, H.; Calero, M.; Rabano, A.; Madden, S.F.; Adamson, K.; Forster, R.; Spain, E.; Prehn, J.H.M.; Henshall, D.C.; et al. Elevated plasma microRNA-206 levels predict cognitive decline and progression to dementia from mild cognitive impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef] [Green Version]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas Analysis Reveals Distinct Extracellular RNA Cargo Types and Their Carriers Present across Human Biofluids. Cell 2019, 177, 463–477.e15. [Google Scholar] [CrossRef] [Green Version]
- Musunuri, S.; Khoonsari, P.E.; Mikus, M.; Wetterhall, M.; Häggmark-Mänberg, A.; Lannfelt, L.; Erlandsson, A.; Bergquist, J.; Ingelsson, M.; Shevchenko, G.; et al. Increased Levels of Extracellular Microvesicle Markers and Decreased Levels of Endocytic/Exocytic Proteins in the Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2016, 54, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Badhwar, A.; Haqqani, A.S. Biomarker potential of brain-secreted extracellular vesicles in blood in Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular vesicle-associated aβ mediates trans-neuronal bioenergetic and ca2+-handling deficits in alzheimer’s disease models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Baker, L.D.; Vitiello, M.V.; Rissman, R.A. Growth Hormone-Releasing Hormone Modulation of Neuronal Exosome Biomarkers in Mild Cognitive Impairment. J. Alzheimer’s Dis. 2018, 66, 971–981. [Google Scholar] [CrossRef]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.J.; Mustapic, M.; Goetz, E.J.; Kapogiannis, D. Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Hum. Brain Mapp. 2017, 38, 1933–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustapic, M.; Tran, J.; Craft, S.; Kapogiannis, D. Extracellular Vesicle Biomarkers Track Cognitive Changes Following Intranasal Insulin in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Nogueras-Ortiz, C.; Mustapic, M.; Mullins, R.J.; Abner, E.L.; Schwartz, J.B.; Kapogiannis, D. Deficient neurotrophic factors of CSPG4-type neural cell exosomes in Alzheimer disease. FASEB J. 2019, 33, 231–238. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Pant, S.; Hilton, H.; Burczynski, M.E. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharmacol. 2012, 83, 1484–1494. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Liu, C.G.; Song, J.; Zhang, Y.Q.; Wang, P.C. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 2395–2400. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal miRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The Serum Exosome Derived MicroRNA−135a, −193b, and −384 Were Potential Alzheimer’s Disease Biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Xu, Y.; Xu, W.; Zhou, Q.; Chen, Q.; Yang, M.; Feng, F.; Liu, Y.; Zhu, X.; Yu, M.; et al. Serum Exosomal miR-223 Serves as a Potential Diagnostic and Prognostic Biomarker for Dementia; Elsevier: Amsterdam, The Netherlands, 2018; Volume 379, ISBN 8651188773631. [Google Scholar]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front. Neurosci. 2019, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.G.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef]
- Rani, A.; O’Shea, A.; Ianov, L.; Cohen, R.A.; Woods, A.J.; Foster, T.C. miRNA in circulating microvesicles as biomarkers for age-related cognitive decline. Front. Aging Neurosci. 2017, 9, 1–10. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell. Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef]
- Chen, J.; Qi, Y.; Liu, C.-F.; Lu, J.-M.; Shi, J.; Shi, Y. MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer’s disease. J. Gene Med. 2018, 20, e3014. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, X.; Liu, C.; Gao, S.; Ping, H.; Wang, J.; Wang, P. MiR-214-3p attenuates cognition defects via the inhibition of autophagy in SAMP8 mouse model of sporadic Alzheimer’s disease. Neurotoxicology 2016, 56, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA Changes in Alzheimer’s Disease Brain and CSF Yields Putative Biomarkers and Insights into Disease Pathways. J. Alzheimer’s Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, S.S.; Nygaard, A.B.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with Alzheimer’s disease and other types of dementia—An exploratory study. Transl. Neurodegener. 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, J.; Xu, J.; Cheng, J.; Jiao, D.; Zhou, C.; Dai, Y.; Chen, Q. Lower serum levels of miR-29c-3p and miR-19b-3p as biomarkers for Alzheimer’s disease. Tohoku J. Exp. Med. 2017, 242, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, N.; Kikuchi, M.; Miyashita, A.; Hatsuta, H.; Saito, Y.; Kasuga, K.; Murayama, S.; Ikeuchi, T.; Kuwano, R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosín-Tomás, M.; Antonell, A.; Lladó, A.; Alcolea, D.; Fortea, J.; Ezquerra, M.; Lleó, A.; Martí, M.J.; Pallàs, M.; Sanchez-Valle, R.; et al. Plasma miR-34a-5p and miR-545-3p as Early Biomarkers of Alzheimer’s Disease: Potential and Limitations. Mol. Neurobiol. 2017, 54, 5550–5562. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, L. Circulating Exosomal miRNA as Diagnostic Biomarkers of Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.-H.; Liu, Y.-N. Downregulated serum miR-223 servers as biomarker in Alzheimer’s disease. Cell Biochem. Funct. 2016, 34, 233–237. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, C.; Sun, A.; Wang, Y.; Zhou, S. Quantification of microRNA-210 in the cerebrospinal fluid and serum: Implications for Alzheimer’s disease. Exp. Ther. Med. 2015, 9, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.G.; Wang, J.L.; Li, L.; Xue, L.X.; Zhang, Y.Q.; Wang, P.C. MicroRNA-135a and -200b, potential Biomarkers for Alzheimer’s disease, regulate β secretase and amyloid precursor protein. Brain Res. 2014, 1583, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Wang, J.; Li, Q.; Ping, H.; Gao, S.; Wang, P. MIR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Reddy, P.H. MicroRNA-455-3p as a Potential Biomarker for Alzheimer’s Disease: An Update. Front. Aging Neurosci. 2018, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Derkow, K.; Rössling, R.; Schipke, C.; Krüger, C.; Bauer, J.; Fähling, M.; Stroux, A.; Schott, E.; Ruprecht, K.; Peters, O.; et al. Distinct expression of the neurotoxic microRNA family let-7 in the cerebrospinal fluid of patients with Alzheimer’s disease. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangla-Valls, A.; Molinuevo, J.L.; Altirriba, J.; Sánchez-Valle, R.; Alcolea, D.; Fortea, J.; Rami, L.; Balasa, M.; Muñoz-García, C.; Ezquerra, M.; et al. CSF microRNA Profiling in Alzheimer’s Disease: A Screening and Validation Study. Mol. Neurobiol. 2017, 54, 6647–6654. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Xin, X.; Kan, P.C.; Yan, Y. MicroRNA-613 regulates the expression of brain-derived neurotrophic factor in Alzheimer’s disease. Biosci. Trends 2016, 10, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Kenny, A.; Jiménez-Mateos, E.M.; Zea-Sevilla, M.A.; Rábano, A.; Gili-Manzanaro, P.; Prehn, J.H.M.; Henshall, D.C.; Ávila, J.; Engel, T.; Hernández, F. Proteins and microRNAs are differentially expressed in tear fluid from patients with Alzheimer’s disease. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Ye, X.; Xiong, Y.; Zhu, H.; Miao, J.; Zhang, W.; Wan, J. The Protective Role of microRNA-200c in Alzheimer’s Disease Pathologies Is Induced by Beta Amyloid-Triggered Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2016, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Tian, N.; Cao, Z.; Zhang, Y. MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2014, 30, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Crooke, A.; Huete-toral, F.; Martı, A.; Colligris, B.; Pintor, J. Ocular disorders and the utility of animal models in the discovery of melatoninergic drugs with therapeutic potential. Expert Opin. Drug Discov. 2012, 7, 989–1001. [Google Scholar] [CrossRef]
- van Wijngaarden, P.; Hadoux, X.; Alwan, M.; Keel, S.; Dirani, M. Emerging ocular biomarkers of Alzheimer disease. Clin. Exp. Ophthalmol. 2017, 45, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Verma, S. Use of ocular biomarkers as a potential tool for early diagnosis of Alzheimer’s disease. Indian J. Ophthalmol. 2020, 68, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloid-β deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5136–5143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- More, S.S.; Vince, R. Hyperspectral imaging signatures detect amyloidopathy in alzheimers mouse retina well before onset of cognitive decline. ACS Chem. Neurosci. 2015, 6, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.E.; Muffat, J.A.; Cherny, R.A.; Moir, R.D.; Ericsson, M.H.; Huang, X.; Mavros, C.; Coccia, J.A.; Faget, K.Y.; Fitch, K.A.; et al. Cytosolic β-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet 2003, 361, 1258–1265. [Google Scholar] [CrossRef]
- Kerbage, C.; Sadowsky, C.H.; Tariot, P.N.; Agronin, M.; Alva, G.; Turner, F.D.; Nilan, D.; Cameron, A.; Cagle, G.D.; Hartung, P.D. Detection of Amyloid β Signature in the Lens and Its Correlation in the Brain to Aid in the Diagnosis of Alzheimer’s Disease. Am. J. Alzheimers Dis. Other Demen. 2015, 30, 738–745. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011, 54, S204–S217. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A.; Syed, A.B. Alzheimer’s disease and the eye. Ophthalmic Physiol. Opt. 2008, 16, S2–S8. [Google Scholar] [CrossRef]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef]
- Esquiva, G.; Hannibal, J. Melanopsin-expressing retinal ganglion cells in aging and disease. Histol. Histopathol. 2019, 34, 1299–1311. [Google Scholar]
- Dutescu, R.M.; Li, Q.X.; Crowston, J.; Masters, C.L.; Baird, P.N.; Culvenor, J.G. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Lee, B.; Woo, J.H.; Jeong, J.B.; Jun, I.; Kim, E.K. APP processing and metabolism in corneal fibroblasts and epithelium as a potential biomarker for Alzheimer’s disease. Exp. Eye Res. 2019, 182, 167–174. [Google Scholar] [CrossRef]
- Csosz, É.; Boross, P.; Csutak, A.; Berta, A.; Tóth, F.; Póliska, S.; Török, Z.; Tozsér, J. Quantitative analysis of proteins in the tear fluid of patients with diabetic retinopathy. J. Proteom. 2012, 75, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Çomoǧlu, S.S.; Güven, H.; Acar, M.; Öztürk, G.; Koçer, B. Tear levels of tumor necrosis factor-alpha in patients with Parkinson’s disease. Neurosci. Lett. 2013, 553, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, S.Z.; Koh, S.K.; Chen, L.; Vaz, C.; Tanavde, V.; Li, X.R.; Beuerman, R.W. In-depth analysis of the human tear proteome. J. Proteom. 2012, 75, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Örnek, N.; Dag, E.; Örnek, K. Corneal sensitivity and tear function in neurodegenerative diseases. Curr. Eye Res. 2015, 40, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Kalló, G.; Emri, M.; Varga, Z.; Ujhelyi, B.; Tozsér, J.; Csutak, A.; Csosz, É. Changes in the chemical barrier composition of tears in Alzheimer’s disease reveal potential tear diagnostic biomarkers. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ship, J.A.; Puckett, S.A. Longitudinal Study on Oral Health in Subjects with Alzheimer’s Disease. J. Am. Geriatr. Soc. 1994, 42, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Ship, J.A.; DeCarli, C.; Friedland, R.P.; Baum, B.J. Diminished submandibular salivary flow in dementia of the Alzheimer Type. J. Gerontol. 1990, 45, 61–66. [Google Scholar] [CrossRef]
- Reuster, T.; Rilke, O.; Oehler, J. High correlation between salivary MHPG and CSF MHPG. Psychopharmacology (Berl) 2002, 162, 415–418. [Google Scholar] [CrossRef]
- Formichi, P.; Battisti, C.; Radi, E.; Federico, A. Cerebrospinal fluid tau, Aß, and phosphorylated tau protein for the diagnosis of Alzheimer’s disease. J. Cell. Physiol. 2006, 208, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermejo-Pareja, F.; Antequera, D.; Vargas, T.; Molina, J.A.; Carro, E. Saliva levels of Abeta1-42 as potential biomarker of Alzheimer’s disease: A pilot study. BMC Neurol. 2010, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-B.; Choi, Y.Y.; Song, W.K.; Song, K.-B. Antibody-based magnetic nanoparticle immunoassay for quantification of Alzheimer’s disease pathogenic factor. J. Biomed. Opt. 2013, 19, 051205. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Shi, J.; Lee, M.; Arnold, L.; Al-Hasan, Y.; Heim, J.; McGeer, P. Salivary beta amyloid protein levels are detectable and differentiate patients with Alzheimer’s disease dementia from normal controls: Preliminary findings. BMC Neurol. 2018, 18, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Guo, J.P.; Kennedy, K.; Mcgeer, E.G.; McGeer, P.L. A method for diagnosing Alzheimer’s disease based on salivary amyloid-β protein 42 levels. J. Alzheimer’s Dis. 2017, 55, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Vianna, C.; Freeman, M.; Davies, P. A polymorphic gene nested within an intron of the tau gene: Implications for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 7751–7756. [Google Scholar] [CrossRef] [Green Version]
- Ashton, N.J.; Ide, M.; Schöll, M.; Blennow, K.; Lovestone, S.; Hye, A.; Zetterberg, H. No association of salivary total tau concentration with Alzheimer’s disease. Neurobiol. Aging 2018, 70, 125–127. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Sui, Y.-T.; Peskind, E.R.; Li, G.; Hwang, H.; Devic, I.; Ginghina, C.; Edgar, J.S.; Pan, C.; Goodlett, D.R.; et al. Salivary Tau Species are Potential Biomarkers of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 299–305. [Google Scholar] [CrossRef]
- Pekeles, H.; Qureshi, H.Y.; Paudel, H.K.; Schipper, H.M.; Gornistky, M.; Chertkow, H. Development and validation of a salivary tau biomarker in Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 53–60. [Google Scholar] [CrossRef]
- Carro, E.; Bartolomé, F.; Bermejo-Pareja, F.; Villarejo-Galende, A.; Molina, J.A.; Ortiz, P.; Calero, M.; Rabano, A.; Cantero, J.L.; Orive, G. Early diagnosis of mild cognitive impairment and Alzheimer’s disease based on salivary lactoferrin. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 8, 131–138. [Google Scholar] [CrossRef]
- Boston, P.F.; Gopalkaje, K.; Manning, L.; Middleton, L.; Loxley, M. Developing a simple laboratory test for Alzheimer’s disease: Measuring acetylcholinesterase in saliva—A pilot study. Int. J. Geriatr. Psychiatry 2008, 23, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, S.; Moghadam, N.B.; Ehsani, M.; Mortazavi, H.; Sabour, S.; Bakhshi, M. Can salivary acetylcholinesterase be a diagnostic biomarker for Alzheimer? J. Clin. Diagn. Res. 2017, 11, ZC58–ZC60. [Google Scholar] [CrossRef] [PubMed]
- García-Blanco, A.; Peña-Bautista, C.; Oger, C.; Vigor, C.; Galano, J.-M.; Durand, T.; Martín-Ibáñez, N.; Baquero, M.; Vento, M.; Cháfer-Pericás, C. Reliable determination of new lipid peroxidation compounds as potential early Alzheimer Disease biomarkers. Talanta 2018, 184, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.M.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hee Lee, S.; Kim, I.; Chul Chung, B. Increased urinary level of oxidized nucleosides in patients with mild-to-moderate Alzheimer’s disease. Clin. Biochem. 2007, 40, 936–938. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Ghanbari, K.; Frey, W.H.; Beheshti, I.; Averback, P.; Hauser, S.L.; Ghanbari, H.A.; Wands, J.R. Characterization of the AD7C-NTP cDNA expression in Alzheimer’s disease and measurement of a 41-kD protein in cerebrospinal fluid. J. Clin. Investig. 1997, 100, 3093–3104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, L.; Li, Y.; Gordon, M.L.; Cai, L.; Wang, Y.; Xing, M.; Cheng, Y. Urine AD7c-NTP predicts amyloid deposition and symptom of agitation in patients with Alzheimer’s disease and mild cognitive impairment. J. Alzheimer’s Dis. 2017, 60, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, C.H.; Li, R.J.; Lin, X.L.; Chen, Y.D.; Gao, H.Q.; Shi, S.L. Accuracy of urinary AD7c-NTP for diagnosing alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 40, 153–159. [Google Scholar] [CrossRef]
- Takae, K.; Hata, J.; Ohara, T.; Yoshida, D.; Shibata, M.; Mukai, N.; Hirakawa, Y.; Kishimoto, H.; Tsuruya, K.; Kitazono, T.; et al. Albuminuria increases the risks for both Alzheimer disease and vascular dementia in community-dwelling Japanese elderly: The hisayama study. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Hong, X.; Li, S.; Zhang, Y.; Zhao, Q.; Du, W.; Wang, Y.; Ni, J. Urine-Based Biomarkers for Alzheimer’s Disease Identified Through Coupling Computational and Experimental Methods. J. Alzheimers Dis. 2018, 65, 421–431. [Google Scholar] [CrossRef]
- Agbemenyah, H.Y.; Agis-Balboa, R.C.; Burkhardt, S.; Delalle, I.; Fischer, A. Insulin growth factor binding protein 7 is a novel target to treat dementia. Neurobiol. Dis. 2014, 62, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Chauhan, A.; Mehta, S.; Mehta, P.; Gu, F.; Chauhan, V. Trichostatin A increases the levels of plasma gelsolin and amyloid beta-protein in a transgenic mouse model of Alzheimer’s disease. Life Sci. 2014, 99, 31–36. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Advantages | Disadvantages | |
|---|---|---|
| CSF | Close relationship with the brain High accuracy in the diagnostic process Ability to test a large number of candidate pathophysiological biomarkers High concentration of the biomarkers | Invasive Clinicians require training Positioned in later disease stages, after blood samples, as a confirmatory diagnostic modality Process less accepted by the population and at the risk of causing harm, anxiety, and fear to the patient |
| Blood | Noninvasive, fast and convenient Inexpensive and reproducible Simple to measure (well-established as part of clinical routines globally) No prior training of the clinicians is required Can be performed in a large variety of settings (primary care, hospitals, patient’s home…) Easy to implement in large populations Ability to test a large number of candidate pathophysiological biomarkers First-step of the multi-stage diagnostic process (identification of patients at the earliest stages of the disease) | Less accurate Presence of very low concentrations of the biomarkers once they have crossed the blood-brain barrier and decreased time window for testing Less consistent results (susceptibility to interference with other components) |
| Other matrices (tears, saliva, and urine) | Extremely noninvasive Repeatable collections Easy, no risk of infection, can be self-collected by the patient Cheap Stress-free | Remarkable lack of validated studies Lack of results replicated in larger, multicenter and longitudinal studies |
| miRNAs | Regulation and Localization | References |
|---|---|---|
| miR-let-7d-5p, miR-let-7g-5p, miR-26b-5p, miR-191-5p | ↓ Blood | [131] |
| miR-125a-5p | ↓ Blood | [128] |
| miR-126-3p, miR-23a-3p, miR-151a-3p | ↓ Blood | [129] |
| miR-135b | ↓ Blood | [132] |
| miR-181a | ↓ Blood | [133] |
| miR-194-5p | ↓ Blood | [134] |
| miR-19b-3p, miR-29c-3p, miR-125b-3p | ↓ Blood | [135] |
| miR-31, miR-93 | ↓ Blood | [99] |
| miR-3613-3p, miR-3916, miR-4772-3p, miR-185-5p, miR-20b-3p | ↓ Blood | [123] |
| miR-501-3p | ↓ Blood | [136] |
| miR-545-3p | ↓ Blood | [137] |
| miR-181c | ↓ Blood, ↓ Brain | [133,138] |
| miR-139-5p, miR-141-3p, miR-150-5p, miR-152-3p, miR-23b-3p, miR-24-3p, miR-338-3p, miR-342-3p, miR-125b-5p, miR-342-5p | ↓ Blood, ↓ CSF | [123] |
| miR-1306-5p | ↓ Blood, ↓ CSF | [122,139] |
| miR-143 | ↓ Blood, ↓ CSF | [99,133] |
| miR-15b | ↓ Blood, ↓ CSF | [131,133] |
| miR-15b-3p | ↓ Blood, ↓ CSF | [122,139] |
| miR-193b | ↓ Blood, ↓ CSF | [121,124] |
| miR-223 | ↓ Blood, ↓ CSF | [125,140] |
| miR-451a | ↓ Blood, ↓ CSF | [128,139] |
| miR-106, miR-107, miR-181 | ↓ Brain | [69] |
| miR-106b | ↓ Brain | [138] |
| miR-137, miR-139, miR-153, miR-183, miR-135, miR-124b | ↓ Brain | [66] |
| miR-15a, miR-19b, miR-26b, miR-330 | ↓ Brain | [138] |
| miR-425 | ↓ Brain | [133] |
| miR-146b | ↓ Brain, ↓ CSF | [133] |
| miR-210 | ↓ Brain, ↓ CSF | [133,141] |
| miR-10, miR-126, miR-127, miR-154, miR-194, miR-195, miR-199a, miR-214, miR-221, miR-338, miR-422b, miR-451, miR-455, miR-497, miR-99a, miR-27a-3p | ↓ CSF | [133] |
| miR-16-2, miR-16-5p, miR-605-5p, mir-9-5p, miR-598, miR-136-3p | ↓ CSF | [139] |
| miR-200b | ↓ CSF | [142] |
| miR-214-3p, miR-299-5p | ↓ CSF | [132,143] |
| miR-29b-3p | ↓ CSF | [123] |
| miR-29c | ↓ CSF | [134] |
| miR-29 | ↓ Blood, ↓ Brain, ↑Brain | [69,131,133] |
| miR-125b | ↓ Blood, ↑ Brain, ↑ CSF | [65,66,123] |
| miR-146a | ↓ Blood, ↑ Brain, ↑ CSF | [69,71,99] |
| miR-26a | ↓ Brain (frontal cortex), ↑ Brain (hippocampus) | [133] |
| miR-3065-5p | ↓ Blood, ↑ Brain | [122,123] |
| let-7i-5p | ↓ Blood, ↑ CSF | [129,134] |
| miR-106a-5p, miR-20-5p, miR-425-5p, miR-18b-5p, miR-582-5p | ↑ Blood | [122] |
| miR-106b-3p, miR-20b-5p, miR-146a-5p, miR-195-5p, niR-497-5p | ↑ Blood | [135] |
| miR-455-3p, miR-4668-5p | ↑ Blood | [144] |
| miR-5001-3p | ↑ Blood | [123] |
| miR-519 | ↑ Blood | [140] |
| miR-548at-5p | ↑ Blood | [123] |
| miR-590-5p | ↑ Blood | [134] |
| miR-101-3p, miR-106b-5p, miR-143-3p, miR-335-5p, miR-361-5p, | ↑ Blood, ↑ CSF | [122] |
| miR-138-5p | ↑ Blood, ↑ CSF | [123] |
| miR-155 | ↑ Blood, ↑ CSF | [71,131] |
| miR-15a-5p | ↑ Blood, ↑ CSF | [122,134] |
| miR-659-5p | ↑ Blood, ↑ CSF | [123] |
| miR-100, miR-145, miR-148a, miR-27, miR-34a, miR-381, miR-422a, miR-423, miR-92 | ↑ Brain | [133] |
| miR-128 | ↑ Brain | [66] |
| miR-34 | ↑ Brain | [69] |
| miR-98 | ↑ Brain | [138] |
| miR-let-7b, miR-let7e | ↑ CSF | [145] |
| miR-let-7f, miR-105, miR-138, miR-141, miR-151, miR-186, miR-191, miR-197, miR-204, miR-205, miR-216, miR-302b, miR-30a-3p, miR-30a-5p, miR-30b, miR-30d, miR-32, miR-345, miR-362, miR-371, miR-374, miR-375, miR-380-3p, miR-429, miR-448, miR-449, miR-494, miR-501, miR-517, miR-518, miR-520, miR-526 | ↑ CSF | [133] |
| miR-20a-5p | ↑ CSF | [122] |
| miR-222 | ↑ CSF | [146] |
| miR-331-5p, miR-485-5p, miR-132-5p | ↑ CSF | [139] |
| miR-613 | ↑ CSF | [147] |
| miR-200b-5p | ↑ Eyes | [148] |
| miR-93-5p | ↑ ↓ Blood, ↑ CSF | [122,135] |
| miR-101 | ↑ Blood, ↓ Brain | [131,138] |
| miR-132, miR-212 | ↑ Blood, ↓ Brain | [126,133] |
| miR-200c | ↑ Blood, ↓ Brain (frontal cortex), ↑ Brain (hippocampus) | [133,149] |
| miR-9 | ↑ Blood, ↓ Brain (frontal cortex, cortex), ↑ Brain (hippocampus), ↑ CSF | [66,71,131,133,138] |
| miR-30e-5p | ↑ Blood, ↑ Brain, ↑ CSF, | [122,133] |
| miR-29a | ↑ Blood, ↓ Brain, ↑ CSF | [73,74,130] |
| miR-206 | ↑ Blood, ↑ CSF, ↑ Eyes | [100,150] |
| miR-142-5p | ↑ Blood, ↓ CSF | [133,134] |
| miR-384 | ↑ Blood, ↓ CSF | [124] |
| miR-135a | ↑ Blood, ↓ CSF, ↑ CSF | [124,133,142] |
| miR-125a | ↓ Brain, ↑ CSF | [66,133] |
| miR-29b | ↓ Blood, ↓ Brain, ↑ CSF | [73,74,131] |
| miR-30c | ↑ Brain (frontal cortex), ↓ Brain (hippocampus), ↑ CSF | [133] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ausó, E.; Gómez-Vicente, V.; Esquiva, G. Biomarkers for Alzheimer’s Disease Early Diagnosis. J. Pers. Med. 2020, 10, 114. https://doi.org/10.3390/jpm10030114
Ausó E, Gómez-Vicente V, Esquiva G. Biomarkers for Alzheimer’s Disease Early Diagnosis. Journal of Personalized Medicine. 2020; 10(3):114. https://doi.org/10.3390/jpm10030114
Chicago/Turabian StyleAusó, Eva, Violeta Gómez-Vicente, and Gema Esquiva. 2020. "Biomarkers for Alzheimer’s Disease Early Diagnosis" Journal of Personalized Medicine 10, no. 3: 114. https://doi.org/10.3390/jpm10030114
APA StyleAusó, E., Gómez-Vicente, V., & Esquiva, G. (2020). Biomarkers for Alzheimer’s Disease Early Diagnosis. Journal of Personalized Medicine, 10(3), 114. https://doi.org/10.3390/jpm10030114