Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry



Abstract
:1. Introduction
2. Materials and Methods
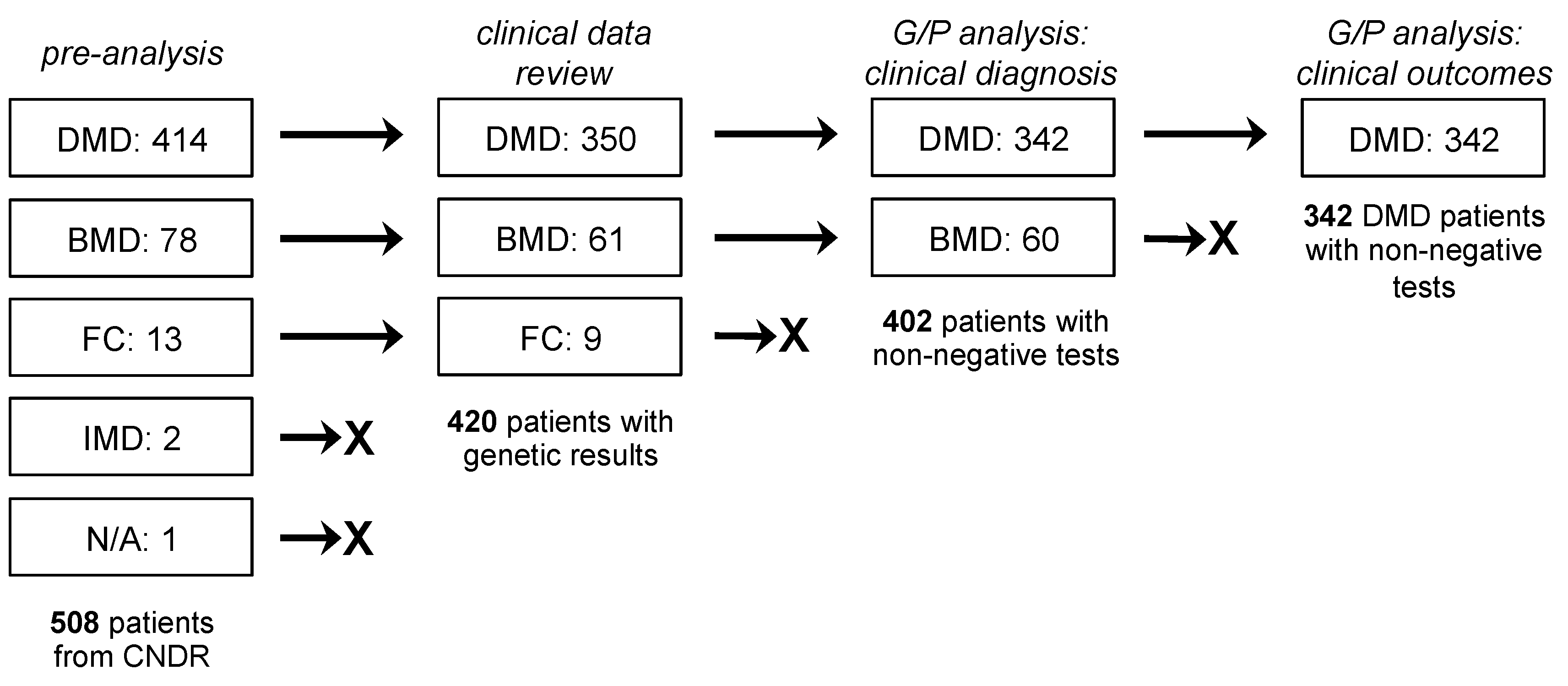
2.1. Study Population and Design
2.2. Statistical Analysis
3. Results
3.1. Clinical Characteristics
3.2. Genetic Characteristics
3.3. Relationships between Genotype and DMD/BMD Diagnosis as Phenotype
3.4. Relationships between Genotype and Clinical Outcome as Phenotype
3.5. Applicability of Exon Skipping Therapy to DMD Patients in Canada
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; Al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [PubMed]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [PubMed]
- Manzur, A.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar]
- Anthony, K.; Cirak, S.; Torelli, S.; Tasca, G.; Feng, L.; Arechavala-Gomeza, V.; Armaroli, A.; Guglieri, M.; Straathof, C.S.; Verschuuren, J.J.; et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: Implications for clinical trials. Brain 2011, 134, 3547–3559. [Google Scholar]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.-F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [PubMed]
- White, S.J.; den Dunnen, J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [PubMed]
- Wei, Y.; McCormick, A.; MacKenzie, A.; O’Ferrall, E.; Venance, S.; Mah, J.K.; Selby, K.; McMillan, H.J.; Smith, G.; Oskoui, M.; et al. The Canadian Neuromuscular Disease Registry: Connecting patients to national and international research opportunities. Paediatr. Child Health 2018, 23, 20–26. [Google Scholar] [PubMed]
- Hodgkinson, V.; Lounsberry, J.; M’Dahoma, S.; Russell, A.; Benstead, T.; Brais, B.; Campbell, C.; Johnston, W.; Lochmüller, H.; McCormick, A.; et al. The Canadian Neuromuscular Disease Registry 2010–2019: A Decade of Facilitating Clinical Research Through a Nationwide, Pan-Neuromuscular Disease Registry. J. Neuromuscul. Dis. 2020, 1–9. [Google Scholar] [CrossRef]
- Mah, J.K.; Selby, K.; Campbell, C.; Nadeau, A.; Tarnopolsky, M.; McCormick, A.; Dooley, J.M.; Kolski, H.; Skalsky, A.J.; Smith, R.G.; et al. A Population-Based Study of Dystrophin Mutations in Canada. Can. J. Neurol. Sci. / J. Can. des Sci. Neurol. 2011, 38, 465–474. [Google Scholar]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [PubMed] [Green Version]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar]
- Tuffery-Giraud, S.; Béroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.P.; Bernard, R.; Cossée, M.; Boisseau, P.; et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [PubMed]
- Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Mahadevappa, M.; Sekar, D.; Purushottam, M.; Thomas, P.T.; Nashi, S.; Nalini, A. Duchenne Muscular Dystrophy and Becker Muscular Dystrophy Confirmed by Multiplex Ligation-Dependent Probe Amplification: Genotype-Phenotype Correlation in a Large Cohort. J. Clin. Neurol. 2017, 13, 91. [Google Scholar] [PubMed]
- Menhart, N. Hybrid spectrin type repeats produced by exon-skipping in dystrophin. Biochim. Biophys. Acta Proteins Proteom. 2006, 1764, 993–999. [Google Scholar]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar] [PubMed]
- Nicolas, A.; Lucchetti-Miganeh, C.; Yaou, R.; Kaplan, J.-C.; Chelly, J.; Leturcq, F.; Barloy-Hubler, F.; Le Rumeur, E. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J. Rare Dis. 2012, 7, 45. [Google Scholar] [PubMed] [Green Version]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar]
- Lim, K.R.Q.; Yokota, T. Invention and Early History of Exon Skipping and Splice Modulation. In Exon Skipping and Inclusion Therapies: Methods and Protocols; Yokota, T., Maruyama, R., Eds.; Springer: New York, NY, USA, 2018; pp. 3–30. [Google Scholar]
- Gibbs, E.M.; Barthélémy, F.; Douine, E.D.; Hardiman, N.C.; Shieh, P.B.; Khanlou, N.; Crosbie, R.H.; Nelson, S.F.; Miceli, M.C. Large in-frame 5′ deletions in DMD associated with mild Duchenne muscular dystrophy: Two case reports and a review of the literature. Neuromuscul. Disord. 2019, 29, 863–873. [Google Scholar]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.-L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar]
- Carsana, A.; Frisso, G.; Tremolaterra, M.R.; Lanzillo, R.; Vitale, D.F.; Santoro, L.; Salvatore, F. Analysis of Dystrophin Gene Deletions Indicates that the Hinge III Region of the Protein Correlates with Disease Severity. Ann. Hum. Genet. 2005, 69, 253–259. [Google Scholar]
- Heydemann, A.; Doherty, K.R.; McNally, E.M. Genetic modifiers of muscular dystrophy: Implications for therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 216–228. [Google Scholar]
- Vo, A.H.; McNally, E.M. Modifier genes and their effect on Duchenne muscular dystrophy. Curr. Opin. Neurol. 2015, 28, 528–534. [Google Scholar] [PubMed] [Green Version]
- Gurvich, O.L.; Maiti, B.; Weiss, R.B.; Aggarwal, G.; Howard, M.T.; Flanigan, K.M. DMD exon 1 truncating point mutations: Amelioration of phenotype by alternative translation initiation in exon 6. Hum. Mutat. 2009, 30, 633–640. [Google Scholar] [PubMed] [Green Version]
- Winnard, A.V.; Mendell, J.R.; Prior, T.W.; Florence, J.; Burghes, A.H. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: Mechanisms of dystrophin production. Am. J. Hum. Genet. 1995, 56, 158–166. [Google Scholar] [PubMed]
- Muntoni, F.; Gobbi, P.; Sewry, C.; Sherratt, T.; Taylor, J.; Sandhu, S.K.; Abbs, S.; Roberts, R.; Hodgson, S.V.; Bobrow, M. Deletions in the 5’ region of dystrophin and resulting phenotypes. J. Med. Genet. 1994, 31, 843–847. [Google Scholar] [PubMed] [Green Version]
- Wang, R.T.; Barthelemy, F.; Martin, A.S.; Douine, E.D.; Eskin, A.; Lucas, A.; Lavigne, J.; Peay, H.; Khanlou, N.; Sweeney, L.; et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum. Mutat. 2018, 39, 1193–1202. [Google Scholar]
- Dwianingsih, E.K.; Malueka, R.G.; Nishida, A.; Itoh, K.; Lee, T.; Yagi, M.; Iijima, K.; Takeshima, Y.; Matsuo, M. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J. Hum. Genet. 2014, 59, 423–429. [Google Scholar]
- Aartsma-Rus, A.; Muntoni, F. 194th ENMC international workshop. 3rd ENMC workshop on exon skipping: Towards clinical application of antisense-mediated exon skipping for Duchenne muscular dystrophy 8–10 December 2012, Naarden, The Netherlands. Neuromuscul. Disord. 2013, 23, 934–944. [Google Scholar]
- Anthony, K.; Arechavala-Gomeza, V.; Ricotti, V.; Torelli, S.; Feng, L.; Janghra, N.; Tasca, G.; Guglieri, M.; Barresi, R.; Armaroli, A.; et al. Biochemical Characterization of Patients With In-Frame or Out-of-Frame DMD Deletions Pertinent to Exon 44 or 45 Skipping. JAMA Neurol. 2014, 71, 32. [Google Scholar]
- Bello, L.; Morgenroth, L.P.; Gordish-Dressman, H.; Hoffman, E.P.; McDonald, C.M.; Cirak, S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 2016, 87, 401–409. [Google Scholar]
- Brogna, C.; Coratti, G.; Pane, M.; Ricotti, V.; Messina, S.; D’Amico, A.; Bruno, C.; Vita, G.; Berardinelli, A.; Mazzone, E.; et al. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS ONE 2019, 14, e0218683. [Google Scholar]
- van den Bergen, J.C.; Ginjaar, H.B.; Niks, E.H.; Aartsma-Rus, A.; Verschuuren, J.J.G.M. Prolonged Ambulation in Duchenne Patients with a Mutation Amenable to Exon 44 Skipping. J. Neuromuscul. Dis. 2014, 1, 91–94. [Google Scholar] [PubMed]
- Suminaga, R.; Takeshima, Y.; Wada, H.; Yagi, M.; Matsuo, M. C-Terminal Truncated Dystrophin Identified in Skeletal Muscle of an Asymptomatic Boy with a Novel Nonsense Mutation of the Dystrophin Gene. Pediatr. Res. 2004, 56, 739–743. [Google Scholar] [PubMed] [Green Version]
- Yue, Y.; Liu, M.; Duan, D. C-Terminal-Truncated Microdystrophin Recruits Dystrobrevin and Syntrophin to the Dystrophin-Associated Glycoprotein Complex and Reduces Muscular Dystrophy in Symptomatic Utrophin/Dystrophin Double-Knockout Mice. Mol. Ther. 2006, 14, 79–87. [Google Scholar] [PubMed]
- Yue, Y.; Li, Z.; Harper, S.Q.; Davisson, R.L.; Chamberlain, J.S.; Duan, D. Microdystrophin Gene Therapy of Cardiomyopathy Restores Dystrophin-Glycoprotein Complex and Improves Sarcolemma Integrity in the Mdx Mouse Heart. Circulation 2003, 108, 1626–1632. [Google Scholar] [PubMed] [Green Version]
- Sweeney, H.L.; Barton, E.R. The dystrophin-associated glycoprotein complex: What parts can you do without? Proc. Natl. Acad. Sci. USA 2000, 97, 13464–13466. [Google Scholar]
- Matsuo, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Kawaguchi, T.; Zhang, Z.; Nishio, H. Dystrophin Dp116: A yet to Be Investigated Product of the Duchenne Muscular Dystrophy Gene. Genes 2017, 8, 251. [Google Scholar]
- Richard, C.A.; Howard, P.L.; D’Souza, V.N.; Klamut, H.J.; Ray, P.N. Cloning and characterization of alternatively spliced isoforms of Dp71. Hum. Mol. Genet. 1995, 4, 1475–1483. [Google Scholar]
- de León, M.B.; Montañez, C.; Gómez, P.; Morales-Lázaro, S.L.; Tapia-Ramírez, V.; Valadez-Graham, V.; Recillas-Targa, F.; Yaffe, D.; Nudel, U.; Cisneros, B. Dystrophin Dp71 Expression Is Down-regulated during Myogenesis. J. Biol. Chem. 2005, 280, 5290–5299. [Google Scholar]
- Kawaguchi, T.; Niba, E.; Rani, A.; Onishi, Y.; Koizumi, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Yoshida, S.; Sakakibara, S.; et al. Detection of Dystrophin Dp71 in Human Skeletal Muscle Using an Automated Capillary Western Assay System. Int. J. Mol. Sci. 2018, 19, 1546. [Google Scholar]
- Yamamoto, T.; Awano, H.; Zhang, Z.; Sakuma, M.; Kitaaki, S.; Matsumoto, M.; Nagai, M.; Sato, I.; Imanishi, T.; Hayashi, N.; et al. Cardiac Dysfunction in Duchenne Muscular Dystrophy Is Less Frequent in Patients With Mutations in the Dystrophin Dp116 Coding Region Than in Other Regions. Circ. Genomic Precis. Med. 2018, 11, e001782. [Google Scholar]
- Lidov, H.G.W.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335. [Google Scholar] [PubMed]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.B.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [PubMed] [Green Version]
- Tandon, A.; Jefferies, J.L.; Villa, C.R.; Hor, K.N.; Wong, B.L.; Ware, S.M.; Gao, Z.; Towbin, J.A.; Mazur, W.; Fleck, R.J.; et al. Dystrophin Genotype–Cardiac Phenotype Correlations in Duchenne and Becker Muscular Dystrophies Using Cardiac Magnetic Resonance Imaging. Am. J. Cardiol. 2015, 115, 967–971. [Google Scholar] [PubMed] [Green Version]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of Dystrophin Deletion Mutations Predicts Age of Cardiomyopathy Onset in Becker Muscular Dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar]
- Ashwath, M.L.; Jacobs, I.B.; Crowe, C.A.; Ashwath, R.C.; Super, D.M.; Bahler, R.C. Left ventricular dysfunction in duchenne muscular dystrophy and genotype. Am. J. Cardiol. 2014, 114, 284–289. [Google Scholar]
- Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491–504. [Google Scholar]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar]
- Béroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.-P.; Voelckel, M.-A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar]
- Waldrop, M.A.; Ben Yaou, R.; Lucas, K.K.; Martin, A.S.; O’Rourke, E.; Ferlini, A.; Muntoni, F.; Leturcq, F.; Tuffery-Giraud, S.; Weiss, R.B.; et al. Clinical Phenotypes of DMD Exon 51 Skip Equivalent Deletions: A Systematic Review. J. Neuromuscul. Dis. 2020, 7, 217–229. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | DMD 1 | BMD 1 | FC 1 | p-Value 2 |
|---|---|---|---|---|
| Number (N) | 350 (83) | 61 (15) | 9 (2) | - |
| Age at visit (yr) | 10.5 (6.8–14.6) | 17.9 (14.1–24.9) | 13.0 (11.9–15.0) | <0.0001 |
| Body mass index | 18.1 (16.2–22.8) | 21.3 (17.6–26.6) | 17.0 (15.4–25.0) | 0.0093 |
| Neuromuscular parameters | ||||
| Wheelchair use | 0.0023 | |||
| >Permanent | 39 (11) | 2 (3) | 0 (0) | |
| >Intermittent | 98 (28) | 10 (16) | 0 (0) | |
| >Never | 156 (45) | 43 (70) | 8 (89) | |
| >Unknown | 57 (16) | 6 (10) | 1 (11) | |
| Can walk without support | 189 (62) | 43 (78) | 9 (100) | 0.0175 |
| Can sit without support | 244 (81) | 52 (95) | 9 (100) | 0.013 |
| Uses steroids | 0.0218 3 | |||
| >Deflazacort | 231 (91) | 6 (67) | 1 (100) | |
| >Prednisone | 19 (7) | 3 (33) | 0 (0) | |
| >Vamorolone | 1 (0) | 0 (0) | 0 (0) | |
| >Testosterone | 3 (1) | 0 (0) | 0 (0) | |
| Cardiac parameters | ||||
| Cardiomyopathy | 37 (11) | 10 (17) | 0 (0) | 0.1928 |
| Age of CM onset (yr) | 13.0 (11.0–14.3) | 23.0 (16.0–33.0) | - | 0.0059 |
| Left ventricle ejection fraction (%) | 63.0 (58.0–68.0) | 60.0 (50.0–65.0) | 68.5 (62.0–70.5) | 0.0325 |
| Uses cardiac medication | 0.0197 4 | |||
| >ACEi/ARB | 69 (70) | 9 (43) | - | |
| >β-blocker | 18 (18) | 5 (24) | - | |
| >Digoxin | 5 (5) | 0 (0) | - | |
| >Statin | 0 (0) | 2 (10) | - | |
| >Antiplatelet | 1 (1) | 1 (5) | - | |
| >Anticoagulant | 0 (0) | 3 (14) | - | |
| >MRA | 3 (3) | 0 (0) | - | |
| Respiratory parameters | ||||
| Uses ventilation assistance | >0.9999 | |||
| >Non-invasive | 30 (9) | 2 (3) | 0 (0) | |
| >Invasive | 1 (0) | 0 (0) | 0 (0) | |
| Forced vital capacity (%) | 76.0 (55.0–93.0) | 88.0 (80.0–100.0) | 74.0 (59.0–85.3) | 0.0018 |
| Sleep apnea | 10 (38) | 0 (0) | 0 (0) | 0.3703 |
| Gastrointestinal parameters | ||||
| Uses feeding tube | 2 (1) | 0 (0) | 0 (0) | >0.9999 |
| Major nutritional route | >0.9999 | |||
| >Oral | 132 (99) | 16 (100) | 2 (100) | |
| >Enteral | 1 (1) | 0 (0) | 0 (0) |
| In-Frame Deletion | Frequency DMD (%) | Frequency BMD (%) | Predicted Repeat Structure 1 |
|---|---|---|---|
| 45–47 | 1 (9.1) | 10 (90.9) | Fractional |
| 45–48 | 0 (0) | 4 (100) | Hybrid |
| 45–49 | 1 (50) | 1 (50) | Fractional |
| 45–51 | 0 (0) | 2 (100) | Hybrid |
| 45–53 | 0 (0) | 1 (100) | Hybrid |
| 45–55 | 0 (0) | 5 (100) | Hybrid |
| 47 | 2 (100) | 0 (0) | Fractional |
| 47–48 | 1 (100) | 0 (0) | Hybrid |
| 48 | 3 (37.5) | 5 (62.5) | Fractional |
| 48–49 | 1 (33.3) | 2 (66.7) | Fractional |
| 48–51 | 2 (100) | 0 (0) | Fractional |
| 49–51 | 1 (50) | 1 (50) | Hybrid |
| 50–51 | 0 (0) | 2 (100) | Fractional |
| 51–52 | 2 (100) | 0 (0) | Fractional |
| Exon/s to Skip | % of DMD Patients with Deletions | % of all DMD Patients | Rank in TREAT-NMD 1 | Rank in Our Population |
|---|---|---|---|---|
| 51 | 17.0 | 12.3 | 1 | 1 |
| 53 | 9.5 | 7.0 | 2 | 4 |
| 45 | 15.8 | 11.1 | 3 | 2 |
| 44 | 12.9 | 9.4 | 4 | 3 |
| 43 | 5.0 | 4.1 | 5 | 7 |
| 46 | 7.9 | 5.8 | 6 | 5 |
| 50 | 5.0 | 3.8 | 7 | 8 |
| 52 | 3.7 | 2.6 | 8 | 10 |
| 55 | 5.0 | 4.7 | 9 | 6 |
| 8 | 2.9 | 3.5 | 10 | 9 |
| 45–55 | 66.8 | 50.9 | n/a | n/a |
| 3–9 | 7.9 | 9.1 | n/a | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241. https://doi.org/10.3390/jpm10040241
Lim KRQ, Nguyen Q, Yokota T. Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. Journal of Personalized Medicine. 2020; 10(4):241. https://doi.org/10.3390/jpm10040241
Chicago/Turabian StyleLim, Kenji Rowel Q., Quynh Nguyen, and Toshifumi Yokota. 2020. "Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry" Journal of Personalized Medicine 10, no. 4: 241. https://doi.org/10.3390/jpm10040241
APA StyleLim, K. R. Q., Nguyen, Q., & Yokota, T. (2020). Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. Journal of Personalized Medicine, 10(4), 241. https://doi.org/10.3390/jpm10040241