A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
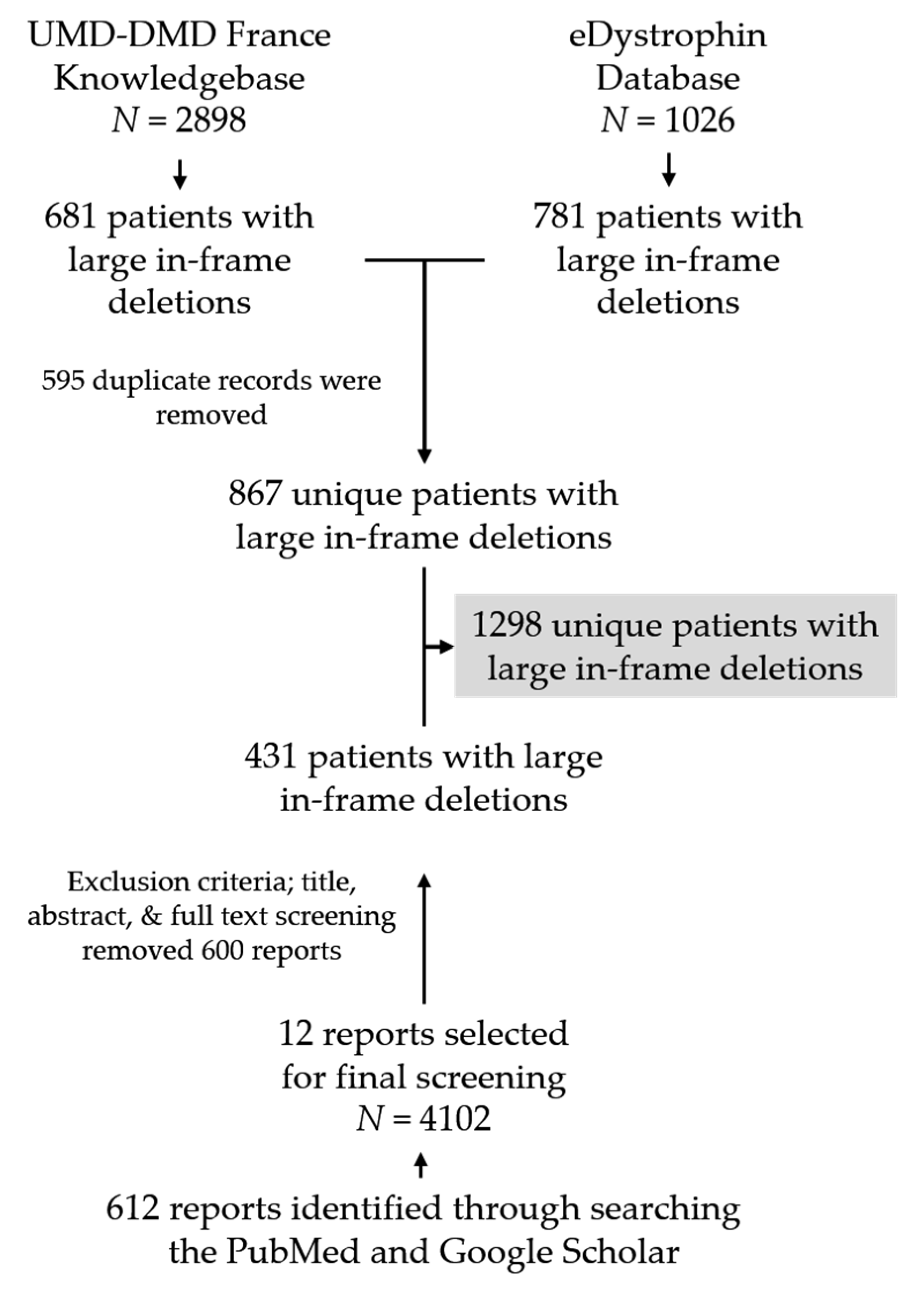
2.1. Clinical Database Search and Inclusion/Exclusion Criteria
2.2. Literature Search and Inclusion/Exclusion Criteria
2.3. Data Extraction and Analysis of the Data
2.4. Exon Skip-Amenable and Exon Skip-Equivalent Mutations
2.5. Statistical Analysis
3. Results
3.1. Patient Pool and Deletional Patterns
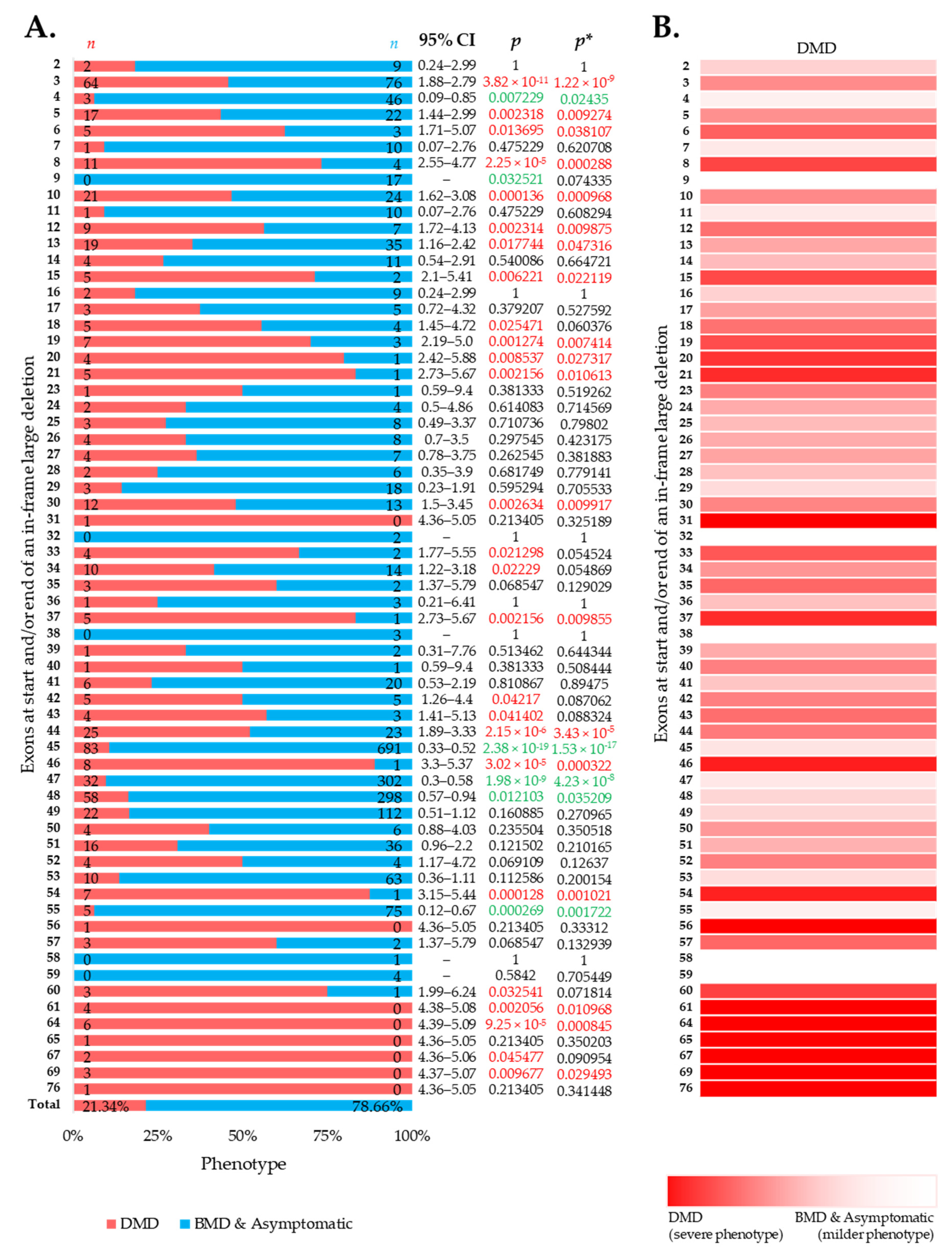
3.2. More BMD and Asymptomatic Phenotypes Were Associated with In-Frame Deletions
3.3. Distribution of Phenotypes for Exon Skip-Amenable Mutations to Each Exon
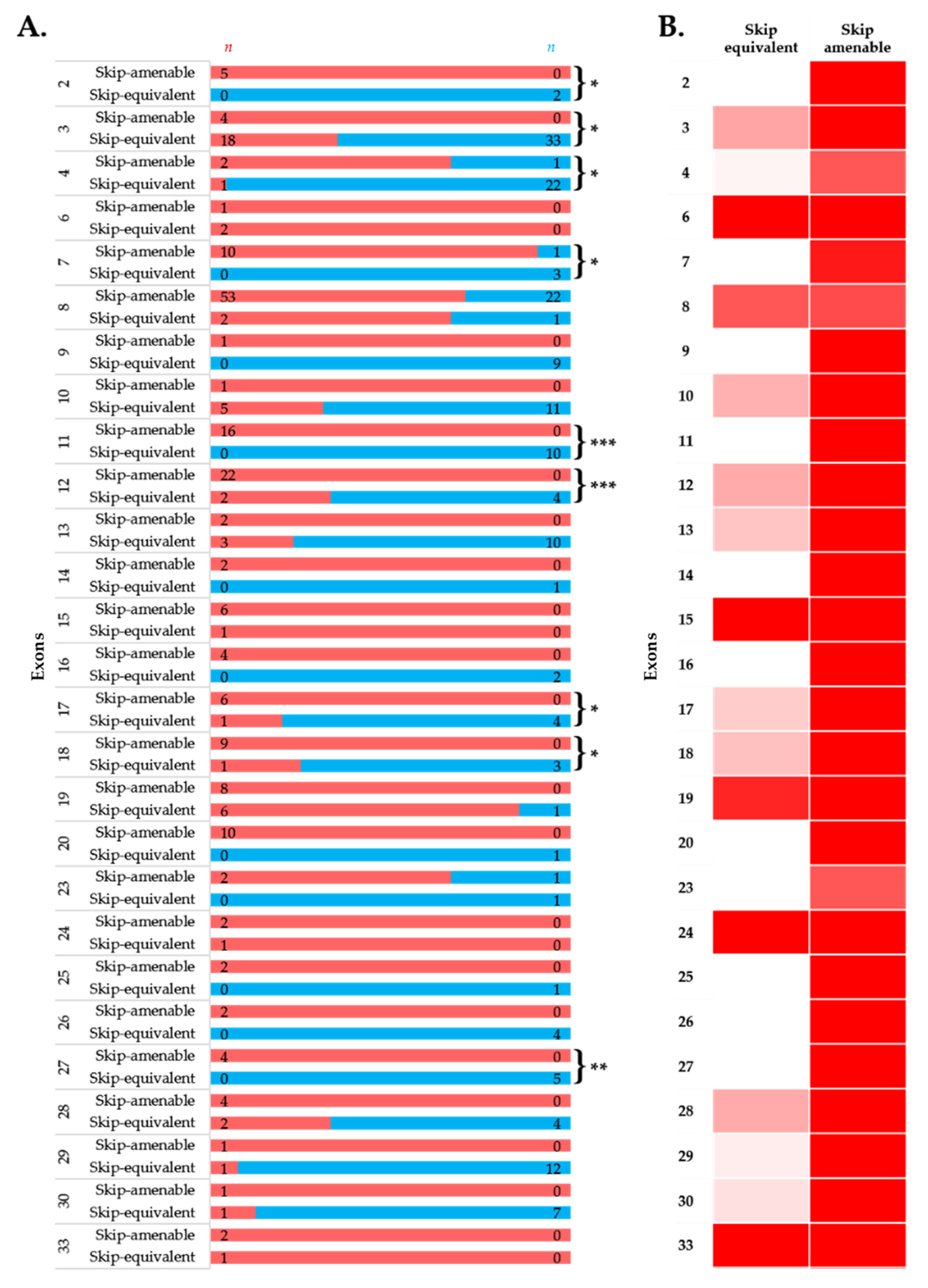
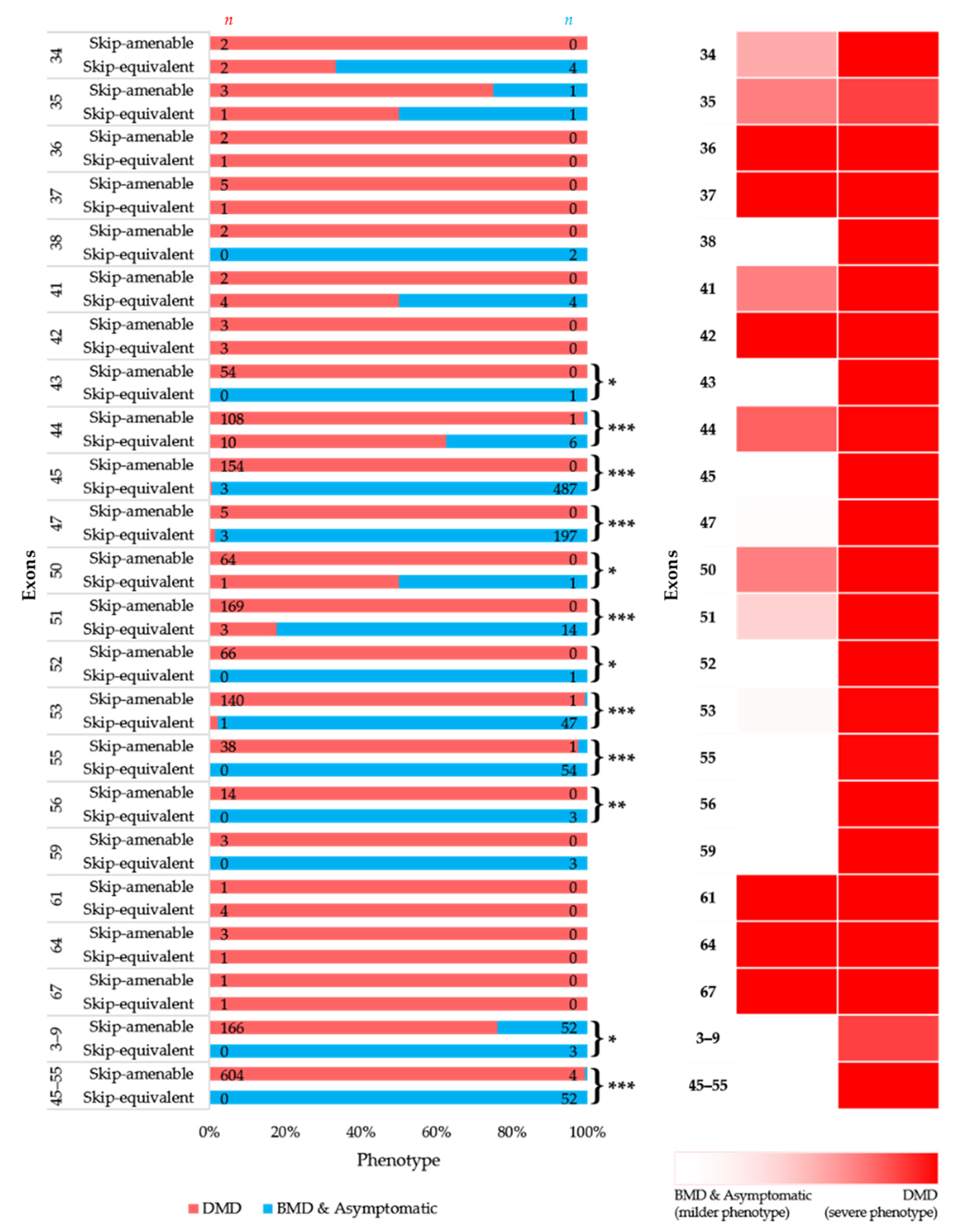
3.4. Comparison of Phenotypes of In-Frame Exon Skip-Equivalent and Out-of-Frame Exon Skip-Amenable Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for duchenne versus becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Roberts, R.G.; Coffey, A.J.; Bobrow, M.; Bentley, D.R. Exon Structure of the Human Dystrophin Gene. Genomics 1993, 16, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Buzin, C.H.; Feng, J.; Yan, J.; Scaringe, W.; Liu, Q.; Den Dunnen, J.; Mendell, J.R.; Sommer, S.S. Mutation rates in the dystrophin gene: A hotspot of mutation at a CpG dinucleotide. Hum. Mutat. 2005, 25, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- White, S.J.; den Dunnen, J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Dunn, D.M.; Von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45–55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, K.; Arechavala-Gomeza, V.; Ricotti, V.; Torelli, S.; Feng, L.; Janghra, N.; Tasca, G.; Guglieri, M.; Barresi, R.; Armaroli, A.; et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. 2014, 71, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bergen, J.C.; Van Westrum, S.M.S.; Dekker, L.; Van Der Kooi, A.J.; De Visser, M.; Wokke, B.H.A.; Straathof, C.S.; Hulsker, M.A.; Aartsma-Rus, A.; Verschuuren, J.J.; et al. Clinical characterisation of Becker muscular dystrophy patients predicts favourable outcome in exon-skipping therapy. J. Neurol. Neurosurg. Psychiatry 2014, 85, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef]
- Tuffery-Giraud, S.; Béroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.P.; Bernard, R.; Cossée, M.; Boisseau, P.; et al. Genotype-phenotype analysis in 2405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef]
- Nicolas, A.; Lucchetti-Miganeh, C.; Yaou, R.B.; Kaplan, J.C.; Chelly, J.; Leturcq, F.; Barloy-Hubler, F.; Le Rumeur, E. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J. Rare Dis. 2012, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North American Association of Central Cancer Registries Protocol for Assessing Duplicate Cases for CiNA, Registry Certification, and Evaluation of 2017 Cases. Available online: https://www.naaccr.org/wp-content/uploads/2018/09/3_Duplicate_protocol2019.pdf (accessed on 20 November 2020).
- Garfinkel, S.L. De-Identification of Personal Information; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2015.
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today (Barc) 2020, 56, 491–504. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.-J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Echigoya, Y.; Nagata, T.; Kuraoka, M.; Kobayashi, M.; Aoki, Y.; Partridge, T.; Maruyama, R.; Takeda, S.; Yokota, T. Efficacy of Multi-exon Skipping Treatment in Duchenne Muscular Dystrophy Dog Model Neonates. Mol. Ther. 2019, 27, 76–86. [Google Scholar] [CrossRef] [Green Version]
- McGauran, N.; Wieseler, B.; Kreis, J.; Schüler, Y.-B.; Kölsch, H.; Kaiser, T. Reporting bias in medical research—A narrative review. Trials 2010, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Yaou, R.B.; Lucas, K.K.; Martin, A.S.; O’Rourke, E.; Ferlini, A.; Ferlini, A.; Muntoni, F.; Leturcq, F.; Tuffery-Giraud, S.; et al. Clinical Phenotypes of DMD Exon 51 Skip Equivalent Deletions: A Systematic Review. J. Neuromuscul. Dis. 2020, 7, 217–229. [Google Scholar] [CrossRef]
- Wang, R.T.; Barthelemy, F.; Martin, A.S.; Douine, E.D.; Eskin, A.; Lucas, A.; Lavigne, J.; Peay, H.; Khanlou, N.; Sweeney, L.; et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum. Mutat. 2018, 39, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Howard, M.T.; Sampson, J.B.; Swoboda, K.J.; Bromberg, M.B.; Mendell, J.R.; Taylor, L.E.; et al. Nonsense mutation-associated Becker muscular dystrophy: Interplay between exon definition and splicing regulatory elements within the DMD gene. Hum. Mutat. 2011, 32, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.-F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q. Molecular dissection of dystrophin identifies the docking site for nNOS. Proc. Natl. Acad. Sci. USA 2013, 110, 387–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Yokota, T. An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [CrossRef]
- Kuntz, N.; Wagner, K.; East, L.; Upadhyay, S.; Han, B.; Koenig, E.; Steiner, D.; Shieh, P. DMD—THERAPY. Neuromuscul. Disord. 2020, 30, S130–S131. [Google Scholar] [CrossRef]
- Wexler, M. Casimersen, Exon 45 Skipping Duchenne Therapy, under FDA Priority Review. Muscular Dystrophy News, 27 August 2020; 4. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, S.; He, M.; Lim, K.R.Q.; Maruyama, R.; Yokota, T. A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. J. Pers. Med. 2021, 11, 46. https://doi.org/10.3390/jpm11010046
Anwar S, He M, Lim KRQ, Maruyama R, Yokota T. A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. Journal of Personalized Medicine. 2021; 11(1):46. https://doi.org/10.3390/jpm11010046
Chicago/Turabian StyleAnwar, Saeed, Merry He, Kenji Rowel Q. Lim, Rika Maruyama, and Toshifumi Yokota. 2021. "A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis" Journal of Personalized Medicine 11, no. 1: 46. https://doi.org/10.3390/jpm11010046
APA StyleAnwar, S., He, M., Lim, K. R. Q., Maruyama, R., & Yokota, T. (2021). A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. Journal of Personalized Medicine, 11(1), 46. https://doi.org/10.3390/jpm11010046