The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients

 , ,
, , 

Abstract
:
1. Introduction
2. Materials and Methods
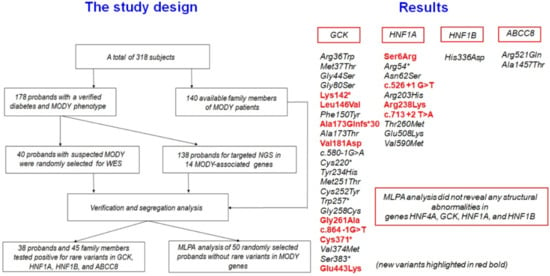
2.1. Patients
2.2. Isolation of Genomic DNA
2.3. Genome Library Preparation and Sequencing
2.4. Bioinformatic Analysis
2.5. Verification Analysis
2.6. MLPA Analysis
3. Results
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steele, A.M.; Shields, B.M.; Wensley, K.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014, 311, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implication of a molecular genetic classification of monogenic β-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213. [Google Scholar] [CrossRef]
- Stanik, J.; Dusatkova, P.; Cinek, O.; Valentinova, L.; Huckova, M.; Skopkova, M.; Dusatkova, L.; Stanikova, D.; Pura, M.; Klimes, I.; et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia 2014, 57, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Yorifuji, T.; Kurokawa, K.; Mamada, M.; Imai, T.; Kawai, M.; Nishi, Y.; Shishido, S.; Hasegawa, Y.; Nakahata, T. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J. Clin. Endocrinol. Metab. 2004, 89, 2905–2908. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; McDonald, T.J.; Ellard, S.; Campbell, M.J.; Hyde, C.; Hattersley, A.T. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 2012, 55, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front. Endocrinol. 2018, 9, 253. [Google Scholar] [CrossRef]
- Shields, B.M.; Hicks, S.; Shepherd, M.H.; Colclough, K.; Hattersley, A.T.; Ellard, S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia 2010, 53, 2504–2508. [Google Scholar] [CrossRef]
- Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T. European Molecular Genetics Quality Network (EMQN) MODY group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 2008, 51, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Fajans, S.S.; Bell, G.I. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Dotto, R.P.; Mathez, A.L.G.; Franco, L.F.; de Sá, J.R.; Weinert, L.S.; Silveiro, S.P.; de Mello Almada Giuffrida, F.; da Silva, M.R.D.; Reis, A.F. Improving the identification of mody mutations by using MLPA technique in the molecular diagnostics routine. Diabetol. Metab. Syndr. 2015, 11, A246. [Google Scholar] [CrossRef] [Green Version]
- Tatsi, E.B.; Kanaka-Gantenbein, C.; Scorilas, A.; Chrousos, G.P.; Sertedaki, A. Next generation sequencing targeted gene panel in Greek MODY patients increases diagnostic accuracy. Pediatr. Diabetes 2020, 21, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int. J. Mol. Sci. 2012, 13, 3245–3276. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Standards of medical care in diabetes-2013. Diabetes Care 2013, 36, 11–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. Cold Spring Harb. Protoc. 2006, 2006, 4455. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Diao, C.; Liu, Y.; Li, M.; Zheng, J.; Zhang, Q.; Yu, M.; Zhang, H.; Ping, F.; Li, M.; et al. Identification and functional analysis of GCK gene mutations in 12 Chinese families with hyperglycemia. J. Diabetes Investig. 2019, 10, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Ivanoshchuk, D.E.; Shakhtshneider, E.V.; Ovsyannikova, A.K.; Mikhailova, S.V.; Rymar, O.D.; Oblaukhova, V.I.; Yurchenko, A.A.; Voevoda, M.I. A rare splice site mutation in the gene encoding glucokinase/hexokinase 4 in a patient with MODY type 2. Vavilov J. Genet. Breed. 2020, 24, 299–305. [Google Scholar] [CrossRef]
- Ovsyannikova, A.K.; Rymar, O.D.; Ivanoshchuk, D.E.; Mikhailova, S.V.; Shakhtshneider, E.V.; Orlov, P.S.; Malakhina, E.S.; Voevoda, M.I. A Case of Maturity Onset Diabetes of the Young (MODY3) in a Family with a Novel HNF1A Gene Mutation in Five Generations. Diabetes Ther. 2018, 9, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovsyannikova, A.K.; Rymar, O.D.; Shakhtshneider, E.V.; Klimontov, V.V.; Koroleva, E.A.; Myakina, N.E.; Voevoda, M.I. ABCC8-Related Maturity-Onset Diabetes of the Young (MODY12): Clinical Features and Treatment Perspective. Diabetes Ther. 2016, 7, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Pedelini, L.; Garcia-Gimeno, M.A.; Marina, A.; Gomez-Zumaquero, J.M.; Rodriguez-Bada, P.; López-Enriquez, S.; Soriguer, F.C.; Cuesta-Muñoz, A.L.; Sanz, P. Structure-function analysis of the alpha5 and the alpha13 helices of human glucokinase: Description of two novel activating mutations. Protein Sci. 2005, 14, 2080–2086. [Google Scholar] [CrossRef] [Green Version]
- Kamata, K.; Mitsuya, M.; Nishimura, T.; Eiki, J.; Nagata, Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004, 12, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Capuano, M.; Garcia-Herrero, C.M.; Tinto, N.; Carluccio, C.; Capobianco, V.; Coto, I.; Cola, A.; Iafusco, D.; Franzese, A.; Zagari, A.; et al. Glucokinase (GCK) mutations and their characterization in MODY2 children of southern Italy. PLoS ONE 2012, 7, e38906. [Google Scholar] [CrossRef] [Green Version]
- Stride, A.; Vaxillaire, M.; Tuomi, T.; Barbetti, F.; Njølstad, P.R.; Hansen, T.; Costa, A.; Conget, I.; Pedersen, O.; Søvik, O.; et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002, 45, 427–435. [Google Scholar] [CrossRef]
- Fenner, D.; Odili, S.; Hong, H.K.; Kobayashi, Y.; Kohsaka, A.; Siepka, S.M.; Vitaterna, M.H.; Chen, P.; Zelent, B.; Grimsby, J.; et al. Generation of N-ethyl-N-nitrosourea (ENU) diabetes models in mice demonstrates genotype-specific action of glucokinase activators. J. Biol. Chem. 2011, 286, 39560–39572. [Google Scholar] [CrossRef] [Green Version]
- Sagen, J.V.; Odili, S.; Bjørkhaug, L.; Zelent, D.; Buettger, C.; Kwagh, J.; Stanley, C.; Dahl-Jørgensen, K.; de Beaufort, C.; Bell, G.I. From clinicogenetic studies of maturity-onset diabetes of the young to unraveling complex mechanisms of glucokinase regulation. Diabetes 2006, 55, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massa, O.; Meschi, F.; Cuesta-Munoz, A.; Caumo, A.; Cerutti, F.; Toni, S.; Cherubini, V.; Guazzarotti, L.; Sulli, N.; Matschinsky, F.M.; et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia 2001, 44, 898–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hager, J.; Blanché, H.; Sun, F.; Vaxillaire, N.V.; Poller, W.; Cohen, D.; Czernichow, P.; Velho, G.; Robert, J.J.; Cohen, N.; et al. Six mutations in the glucokinase gene identified in MODY by using a nonradioactive sensitive screening technique. Diabetes 1994, 43, 730–733. [Google Scholar] [CrossRef]
- Stoffel, M.; Froguel, P.; Takeda, J.; Zouali, H.; Vionnet, N.; Nishi, S.; Weber, I.T.; Harrison, R.W.; Pilkis, S.J.; Lesage, S.; et al. Human glucokinase gene: Isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proc. Natl. Acad. Sci. USA 1992, 89, 7698–7702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellard, S.; Beards, F.; Allen, L.; Shepherd, M.; Ballantyne, E.; Harvey, R.; Hattersley, A.T. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia 2000, 43, 250–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruhova, S.; Dusatkova, P.; Sumnik, Z.; Kolouskova, S.; Pedersen, O.; Hansen, T.; Cinek, O.; Lebl, J. Glucokinase diabetes in 103 families from a country-based study in the Czech Republic: Geographically restricted distribution of two prevalent GCK mutations. Pediatr. Diabetes 2010, 11, 529–535. [Google Scholar] [CrossRef]
- Aloi, C.; Salina, A.; Minuto, N.; Tallone, R.; Lugani, F.; Mascagni, A.; Mazza, O.; Cassanello, M.; Maghnie, M.; d’Annunzio, G. Glucokinase mutations in pediatric patients with impaired fasting glucose. Acta Diabetol. 2017, 54, 913–923. [Google Scholar] [CrossRef]
- George, D.C.; Chakraborty, C.; Haneef, S.A.; Nagasundaram, N.; Chen, L.; Zhu, H. Evolution- and structure-based computational strategy reveals the impact of deleterious missense mutations on MODY 2 (maturity-onset diabetes of the young, type 2). Theranostics 2014, 4, 366–385. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Horikawa, Y.; Enya, M.; Takeda, J.; Imai, Y.; Imai, Y.; Handa, H.; Imai, T. L-Arginine prevents cereblon-mediated ubiquitination of glucokinase and stimulates glucose-6-phosphate production in pancreatic β-cells. Commun. Biol. 2020, 3, 497. [Google Scholar] [CrossRef]
- Boj, S.F.; Parrizas, M.; Maestro, M.A.; Ferrer, J. A transcription factor regulatory circuit in differentiated pancreatic cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14481–14486. [Google Scholar] [CrossRef] [Green Version]
- Reznik, Y.; Dao, T.; Coutant, R.; Chiche, L.; Jeannot, E.; Clauin, S.; Rousselot, P.; Fabre, M.; Oberti, F.; Fatome, A.; et al. Hepatocyte nuclear factor-1 alpha gene inactivation: Cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J. Clin. Endocrinol. Metab. 2004, 89, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Sayer, J.A.; Quinton, R.; Walker, M.; Ellard, S.; Goodship, T.H.J. Monogenic diabetes, renal dysplasia and hypopituitarism: A patient with a HNF1A mutation. QJM Int. J. Med. 2011, 104, 881–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajans, S.S.; Bell, G.I. Phenotypic heterogeneity between different mutations of MODY subtypes and within MODY pedigrees. Diabetologia 2006, 49, 1106–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colclough, K.; Bellanne-Chantelot, C.; Saint-Martin, C.; Flanagan, S.E.; Ellard, S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum. Mutat. 2013, 34, 669–685. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, C.; Yin, D.; Zhao, F.; Bao, Z.; Zhao, Y.; Wang, X.; Li, W.; Wang, T.; Jin, Y.; et al. A Variation in the ABCC8 Gene Is Associated with Type 2 Diabetes Mellitus and Repaglinide Efficacy in Chinese Type 2 Diabetes Mellitus Patients. Intern. Med. 2019, 58, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Vaxillaire, M.; Dechaume, A.; Busiah, K.; Cavé, H.; Pereira, S.; Scharfmann, R.; de Nanclares, G.P.; Castano, L.; Froguel, P.; Polak, M. SUR1-Neonatal Diabetes Study Group. New ABCC8 mutations in relapsing neonatal diabetes and clinical features. Diabetes 2007, 56, 1737–1741. [Google Scholar] [CrossRef] [Green Version]
- Edghill, E.L.; Flanagan, S.E.; Ellard, S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef]
- Calabria, A.C.; Li, C.; Gallagher, P.R.; Stanley, C.A.; De León, D.D. GLP-1 receptor antagonist exendin-(9–39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes 2012, 61, 2585–2591. [Google Scholar] [CrossRef] [Green Version]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; Sanyoura, M.; Greeley, S.A.W.; et al. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef] [Green Version]
- El-Khairi, R.; Vallier, L. The role of hepatocyte nuclear factor 1β in disease and development. Diabetes Obes. Metab. 2016, 18, 23–32. [Google Scholar] [CrossRef]
- Klimontov, V.V.; Bulumbaeva, D.M.; Koroleva, E.A.; Ovsyannikova, A.K.; Rymar, O.D.; Ivanoshchuk, D.E.; Shakhtshneider, E.V. Maturity-onset diabetes of the young due to HNF1B mutation: A case report. In Proceedings of the Systems Biology and Biomedicine, SBioMed-2018\Systems Biology (BGRS\SB-2018), Novosibirsk, Russia, 20–25 August 2018; p. 64, ISBN 978-5-91291-040-1. [Google Scholar]
- Weber, S.; Moriniere, V.; Knüppel, T.; Charbit, M.; Dusek, J.; Ghiggeri, G.M.; Jankauskiené, A.; Mir, S.; Montini, G.; Peco-Antic, A.; et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J. Am. Soc. Nephrol. 2006, 17, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Karges, B.; Bergmann, C.; Scholl, K.; Heinze, E.; Rasche, F.M.; Zerres, K.; Debatin, K.M.; Wabitsch, M.; Karges, W. Digenic Inheritance of Hepatocyte Nuclear Factor-1α and -1β With Maturity-Onset Diabetes of the Young, Polycystic Thyroid, and Urogenital Malformations. Diabetes Care 2007, 30, 1613–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, I.; Martínez, R.; Rica, I.; Martínez de LaPiscina, I.; García-Castaño, A.; Aguayo, A.; Calvo, B.; Castaño, L. Spanish Pediatric Diabetes Collaborative Group. Negative autoimmunity in a Spanish pediatric cohort suspected of type 1 diabetes, could it be monogenic diabetes? PLoS ONE 2019, 14, e0220634. [Google Scholar] [CrossRef] [Green Version]
- Dedov, I.I.; Zubkova, N.A.; Arbatskaya, N.Y.; Akopova, A.G.; Tyul’pakov, A.N. MODY2: Clinical and molecular genetic characteristics of 13 cases of the disease. The first description of MODY in Russia. Probl. Endokrinol. 2009, 55, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Glotov, O.S.; Serebryakova, E.A.; Turkunova, M.E.; Efimova, O.A.; Glotov, A.S.; Barbitoff, Y.A.; Nasykhova, Y.A.; Predeus, A.V.; Polev, D.E.; Fedyakov, M.A.; et al. Whole-exome sequencing in Russian children with non-type 1 diabetes mellitus reveals a wide spectrum of genetic variants in MODY-related and unrelated genes. Mol. Med. Rep. 2019, 20, 4905–4914. [Google Scholar] [CrossRef] [Green Version]
- GoodSmith, M.S.; Skandari, M.R.; Huang, E.S.; Naylor, R.N. The Impact of Biomarker Screening and Cascade Genetic Testing on the Cost-Effectiveness of MODY Genetic Testing. Diabetes Care 2019, 42, 2247–2255. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MODY Type | Gene | Protein Name | Genomic Location GRCh37 (hg19) |
|---|---|---|---|
| MODY1 | HNF4A | Hepatic Nuclear Factor 4 Alpha | chr20:42,984,340-43,061,485 |
| MODY2 | GCK | Glucokinase (Hexokinase 4) | chr7:44,183,870-44,237,769 |
| MODY3 | HNF1A | Hepatocyte Nuclear Factor 1-Alpha | chr12:121,416,346-121,440,315 |
| MODY4 | PDX1 | Pancreatic and Duodenal Homeobox 1 | chr13:28,494,157-28,500,451 |
| MODY5 | HNF1B | Hepatocyte Nuclear Factor 1-Beta | chr17:36,046,434-36,105,237 |
| MODY6 | NEUROD1 | Neuronal Differentiation 1 | chr2:182,537,815-182,545,603 |
| MODY7 | KLF11 | Krüppel-Like Factor 11 | chr2:10,182,976-10,194,963 |
| MODY8 | CEL | Carboxyl Ester Lipase | chr9:135,937,365-135,947,248 |
| MODY9 | PAX4 | Paired Box 4 | chr7:127,250,346-127,255,982 |
| MODY10 | INS | Insulin | chr11:2,181,009-2,182,571 |
| MODY11 | BLK | BLK Proto-Oncogene, Src Family Tyrosine Kinase | chr8:11,351,510-11,422,113 |
| MODY12 | ABCC8 | ATP-Binding Cassette Subfamily C Member 8 | chr11:17,414,432-17,498,449 |
| MODY13 | KCNJ11 | Potassium Inwardly Rectifying Channel Subfamily J Member 11 | chr11:17,406,795-17,410,878 |
| MODY14 | APPL1 | Adaptor Protein, Phosphotyrosine Interacting with PH Domain and Leucine Zipper 1 | chr3:57,261,765-57,307,499 |
| Patient ID, Gender | Gene | Variant Status | Nucleotide Changes * | Amino Acid Changes | Location | Genotype | Minor Allele Frequency (gnomAD) | dbSNP ID | ClinVar Variation ID | HGMD | Pathogenicity According to ACMG [21], Evidence | LOVD Database ID | Segregation with Phenotype in Family |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P6, Female | GCK | Known | c.106C>T | p.Arg36Trp | Exon 2 | Heterozygous | 0.000014 | rs762263694 | 431973 | CM940823 | PR | GCK_000007 | Yes |
| P50, Male | Heterozygous | Yes | |||||||||||
| P59, Female | GCK | Known | c.110T>C | p.Met37Thr | Exon 2 | Heterozygous | NA | NA | NA | NA | PR | GCK_000100 | Yes |
| P17, Female | GCK | Known | c.130G>A | p.Gly44Ser | Exon 2 | Heterozygous | NA | rs267601516 | 76898 | CM013265 | PR | GCK_000029 | Yes |
| P74, Female | GCK | Known | c.238G>A | p.Gly80Ser | Exon 3 | Heterozygous | NA | rs1554335761 | 449415 | CM970630 | PR | NA | Yes |
| P51, Male | GCK | Novel | c.424A>T | p.Lys142* | Exon 4 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PM2, PM1, PP1 | NA | Yes |
| P4, Male | GCK | Novel | c.436C>G | p. Leu146Val | Exon 4 | Heterozygous | NA | NA | NA | NA | Pathogenic, PS1, PS3, PM2, PP1 | NA | Yes |
| P10, Male | GCK | Known | c.449T>A | p. Phe150Tyr | Exon 4 | Heterozygous | NA | rs193922297 | 129142 | CM097114 | PR | NA | Yes |
| P86, Female | GCK | Novel | c.517_520del | p.Ala173Glnfs*30 | Exon 5 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PS1, PM2, PM4, PP1, PP3 | NA | Yes |
| P90, Male | GCK | Known | c.517G>A | p.Ala173Thr | Exon 5 | Heterozygous | NA | NA | NA | NA | Pathogenic, PS1, PM2, PM5, PP1, PP3 | GCK_000217 | Yes |
| P80, Female | GCK | Novel | c.542T>A | Val181Asp | Exon 5 | Heterozygous | NA | NA | NA | NA | Pathogenic, PS1, PM2, PM5, PP1, PP3 | NA | Yes |
| P46, Female | Heterozygous | Yes | |||||||||||
| P14, Female | GCK | Known | c.580-1G>A | - | Intron 5 | Heterozygous | NA | rs1554335421 | 449414 | CS052048 | PR | NA | Yes |
| P30, Female | GCK | Known | c.660C>A | p.Сys220* | Exon 6 | Heterozygous | NA | NA | NA | CM020443 | PR | NA | Yes |
| P68, Female | GCK | Known | c.700T>C | p.Tyr234His | Exon 7 | Heterozygous | NA | NA | NA | CM096864 | PR | NA | Yes |
| P40, Female | GCK | Known | c.752T>C | p. Мet251Тhr | Exon 7 | Heterozygous | NA | rs193922326 | 36251 | CM096876 | PR | NA | Yes |
| P67, Male | GCK | Known | c.755G>A | p.Сys252Tyr | Exon 7 | Heterozygous | NA | NA | NA | CM021266 | PR | NA | NA |
| P83, Male | GCK | Known | c.771G>A | p.Trp257* | Exon7 | Heterozygous | NA | NA | NA | NA | Reported in [22] | NA | NA |
| P3, Male | GCK | Known | c.772G>T | p.Gly258Cys | Exon 7 | Heterozygous | NA | rs1583596378 | 804857 | CM032578 | PR | NA | NA |
| P77, Female | Yes | ||||||||||||
| P88, Female | GCK | Novel | с.782G>C | p.Gly261Ala | Exon 7 | Heterozygous | NA | NA | NA | NA | Pathogenic, PS1, PM2, PM5, PP3 | NA | NA |
| P54, Male | GCK | Novel | c.864-1G>T | - | Intron 7 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PM2, PP1, PP3 | NA | Yes |
| P57, Male | GCK | Novel | c.1113C>A | p.Cys371* | Exon 9 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PS1, PM1, PM2, PM5, PP1 | NA | Yes |
| P70, Female | GCK | Known | c.1120G>A | p.Val374Met | Exon 9 | Heterozygous | NA | rs1415041911 | 447380 | CM096927 | PR | NA | Yes |
| P61, Female | GCK | Known | c.1148C>A | p.Ser383* | Exon 9 | Heterozygous | 0.0000042 | rs777870079 | NA | CM032579 | PR | NA | Yes |
| P87, Male | GCK | Novel | c.1327G>A | p.Glu443Lys | Exon 10 | Heterozygous | NA | NA | NA | NA | Likely pathogenic, PM1, PM2, PP2, PP3 | NA | NA |
| P19, Female | HNF1A | Novel | c.18C>G | p.Ser6Arg | Exon 1 | Heterozygous | NA | NA | NA | NA | Likely pathogenic, PS4, PM2, PP1, PP2, PP3 | NA | Yes |
| P11, Female | HNF1A | Known | c.185A>G | p.Asn62Ser | Exon 1 | Heterozygous | 0.00012 | rs377129682 | 447485 | CM064300 | PR | HNF1A_000235 | NA |
| P34, Female | HNF1A | Novel | c.526 +1G>T | - | Intron 2 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PM2, PP1, PP2, PP3 | NA | Yes |
| P65, Female | HNF1A | Known | c.608G>A | p.Arg203His | Exon 3 | Heterozygous | 0.000008 | rs587780357 | 129235 | CM993816 | PR | HNF1A_000137 | Yes |
| P91, Female | HNF1A | Novel | c.713G>A | p.Arg238Lys | Exon 3 | Heterozygous | NA | NA | NA | NA | Likely pathogenic, PS1, PM2, PP2, PP3 | NA | NA |
| P78, Male | HNF1A | Novel | c.713 +2T>A | - | Intron 3 | Heterozygous | NA | NA | NA | NA | Pathogenic, PVS1, PM2, PP1, PP2, PP3 | NA | Yes |
| P82, Female | HNF1A | Known | c.779C>T | p.Thr260Met | Exon 4 | Heterozygous | 0.0000040 | rs886039544 | 265436 | CM971457 | PR | HNF1A_000148 | NA |
| P16, Female | HNF1A | Known | c.1522G > A | p.Glu508Lys | Exon 6 | Heterozygous | 0.00044 | rs483353044 | 135665 | CM082841 | PR | HNF1A_000214 | NA |
| P81, Male | HNF1A | Known | c.1768G>A | p.Val590Met | Exon 9 | Heterozygous | 0.0000042 | rs1168108747 | 447484 | NA | Uncertain significance, PM2, PP1, PP2, PP3 | NA | Yes |
| P73, Male | HNF1A | Known | c.160C>T | p.Arg54* | Exon 1 | Heterozygous | 0.0000042 | rs766956862 | 805632 | CM032035 | PR | HNF1A_000220 | Yes |
| ABCC8 | Known | c.1562G>A | p.Arg521Gln | Exon 10 | Heterozygous | 0.000095 | rs368114790 | 157683 | CM1212138 | PR | ABCC8_000172 | ||
| P12, Male | ABCC8 | Known | c.4369G>C | p.Ala1457Thr | Exon 36 | Heterozygous | NA | rs72559717 | NA | CM011260 | PR | NA | Yes |
| P27, Female | HNF1B | Known | c.1006C>G | p.His336Asp | Exon 4 | Heterozygous | 0.0002 | rs138986885 | 595653 | CM067046 | PR | HNF1B_000125 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanoshchuk, D.E.; Shakhtshneider, E.V.; Rymar, O.D.; Ovsyannikova, A.K.; Mikhailova, S.V.; Fishman, V.S.; Valeev, E.S.; Orlov, P.S.; Voevoda, M.I. The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients. J. Pers. Med. 2021, 11, 57. https://doi.org/10.3390/jpm11010057
Ivanoshchuk DE, Shakhtshneider EV, Rymar OD, Ovsyannikova AK, Mikhailova SV, Fishman VS, Valeev ES, Orlov PS, Voevoda MI. The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients. Journal of Personalized Medicine. 2021; 11(1):57. https://doi.org/10.3390/jpm11010057
Chicago/Turabian StyleIvanoshchuk, Dinara E., Elena V. Shakhtshneider, Oksana D. Rymar, Alla K. Ovsyannikova, Svetlana V. Mikhailova, Veniamin S. Fishman, Emil S. Valeev, Pavel S. Orlov, and Mikhail I. Voevoda. 2021. "The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients" Journal of Personalized Medicine 11, no. 1: 57. https://doi.org/10.3390/jpm11010057
APA StyleIvanoshchuk, D. E., Shakhtshneider, E. V., Rymar, O. D., Ovsyannikova, A. K., Mikhailova, S. V., Fishman, V. S., Valeev, E. S., Orlov, P. S., & Voevoda, M. I. (2021). The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients. Journal of Personalized Medicine, 11(1), 57. https://doi.org/10.3390/jpm11010057