
A Comprehensive Evaluation of Cross-Omics Blood-Based Biomarkers for Neuropsychiatric Disorders

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Pre-Processing
2.2. Summary-Based and 2-Sample Mendelian Randomization (SMR and 2SMR)
2.3. Simulation Analysis
2.4. Published Transcriptome and Methylome Data Analysis
2.5. Diagnostic Model Construction
2.6. Predictive Model Construction
3. Results
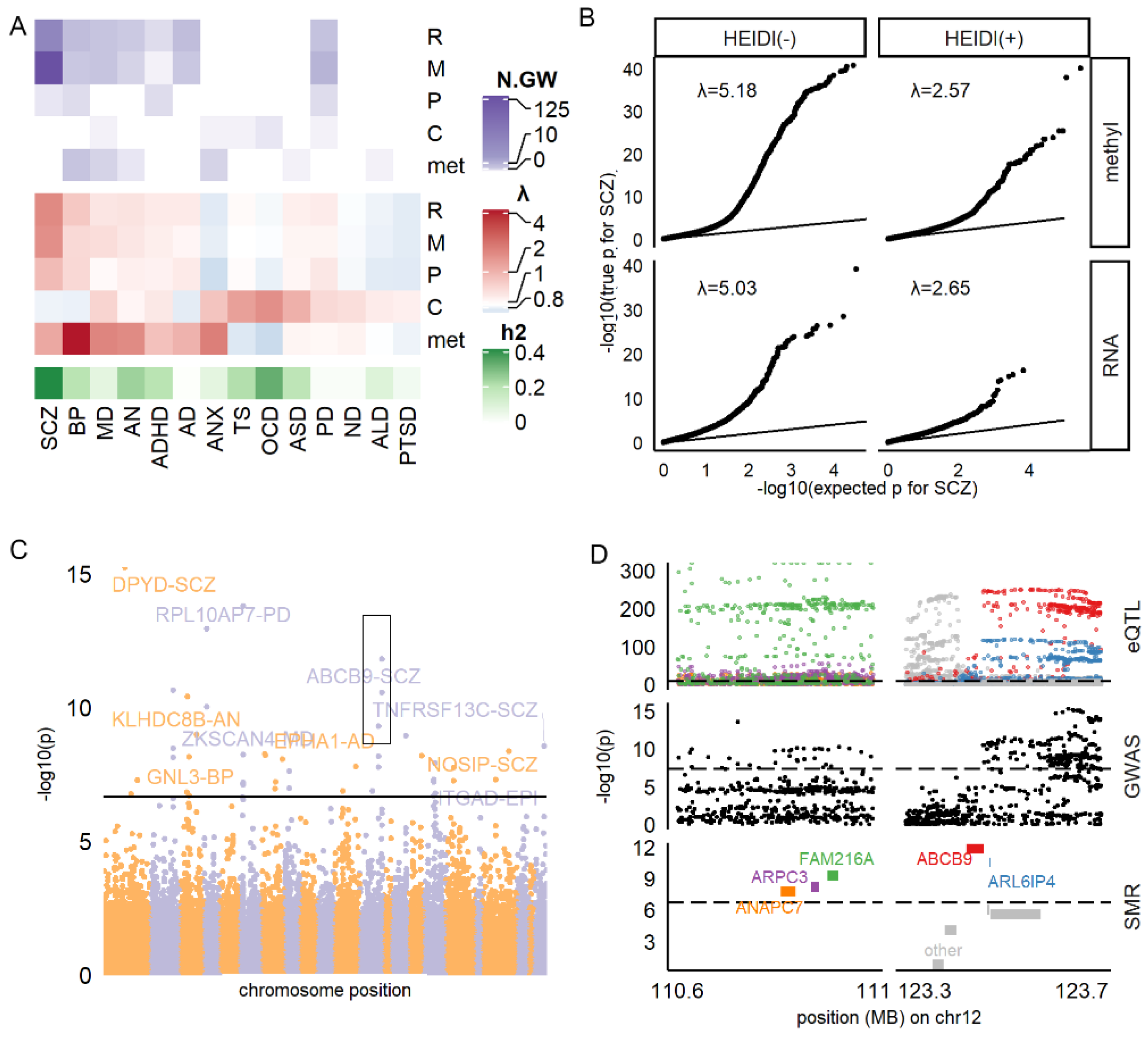
3.1. Identifying All Potential Blood-Based Biomarkers Associated with Brain Disorders
3.2. RNA and Methylation Levels Showing Strong Associations with SCZ, PD, and AD
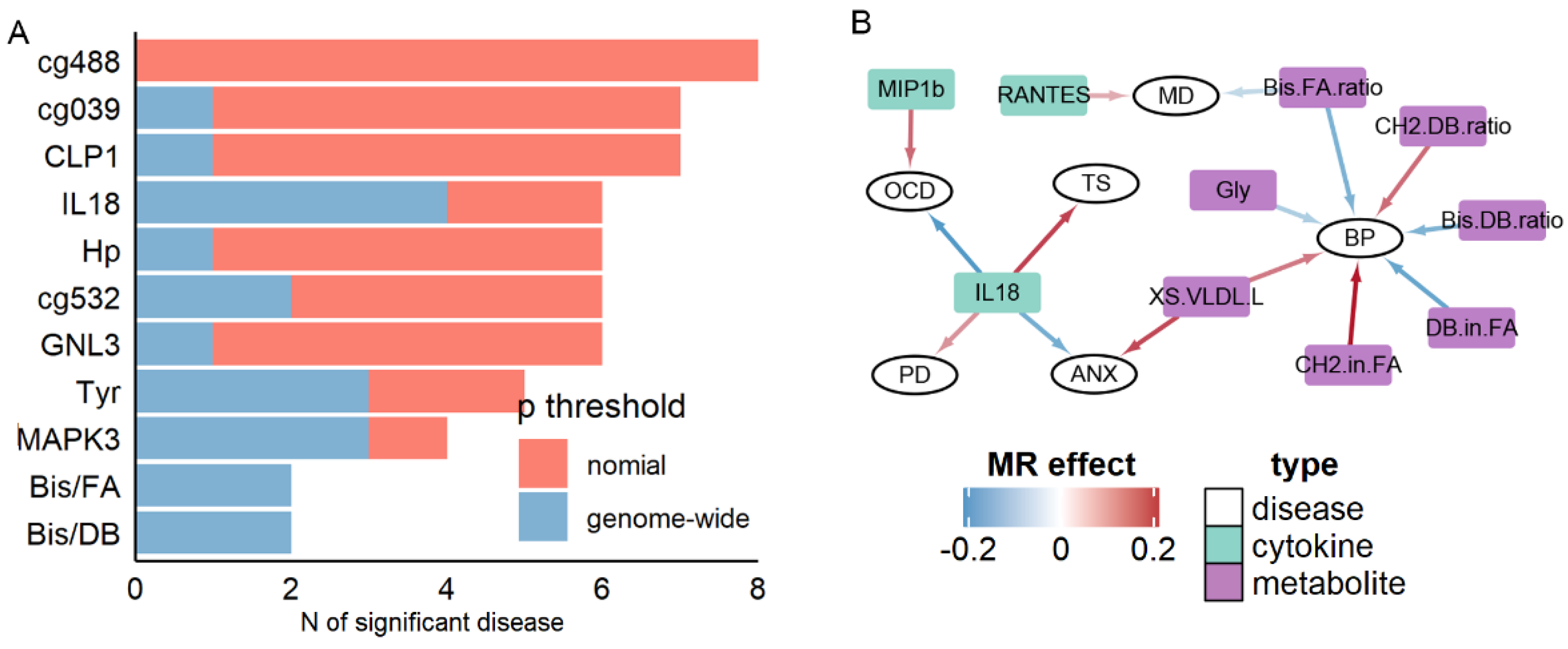
3.3. Cytokines and Metabolites Exhibiting High Pleiotropy
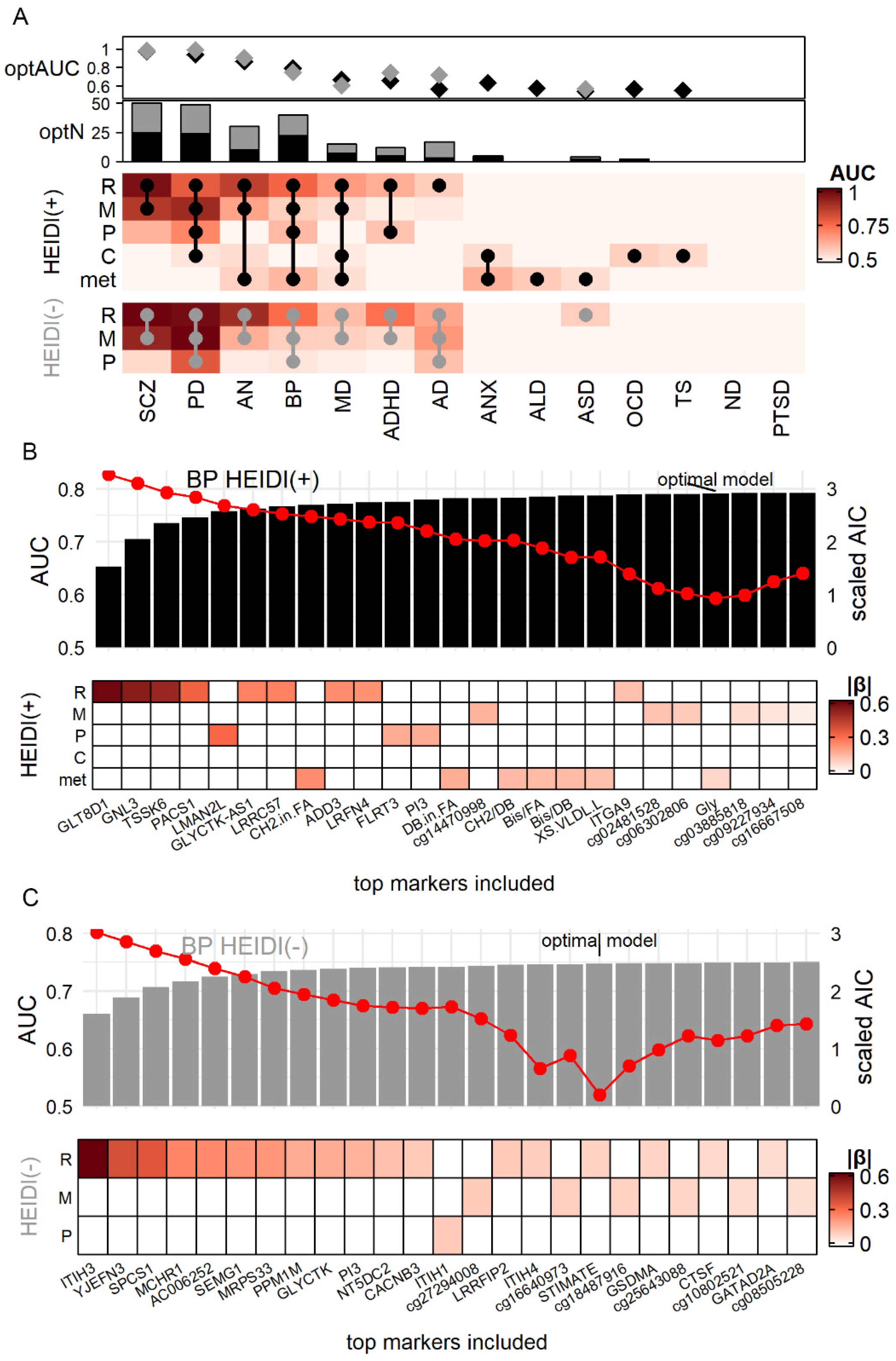
3.4. Simulation Demonstrated the Advantage of Using Cross-Omics Biomarker Combinations
3.5. HEIDI(+) and HEIDI(−) Markers Having Comparable Power in Real-World Validation
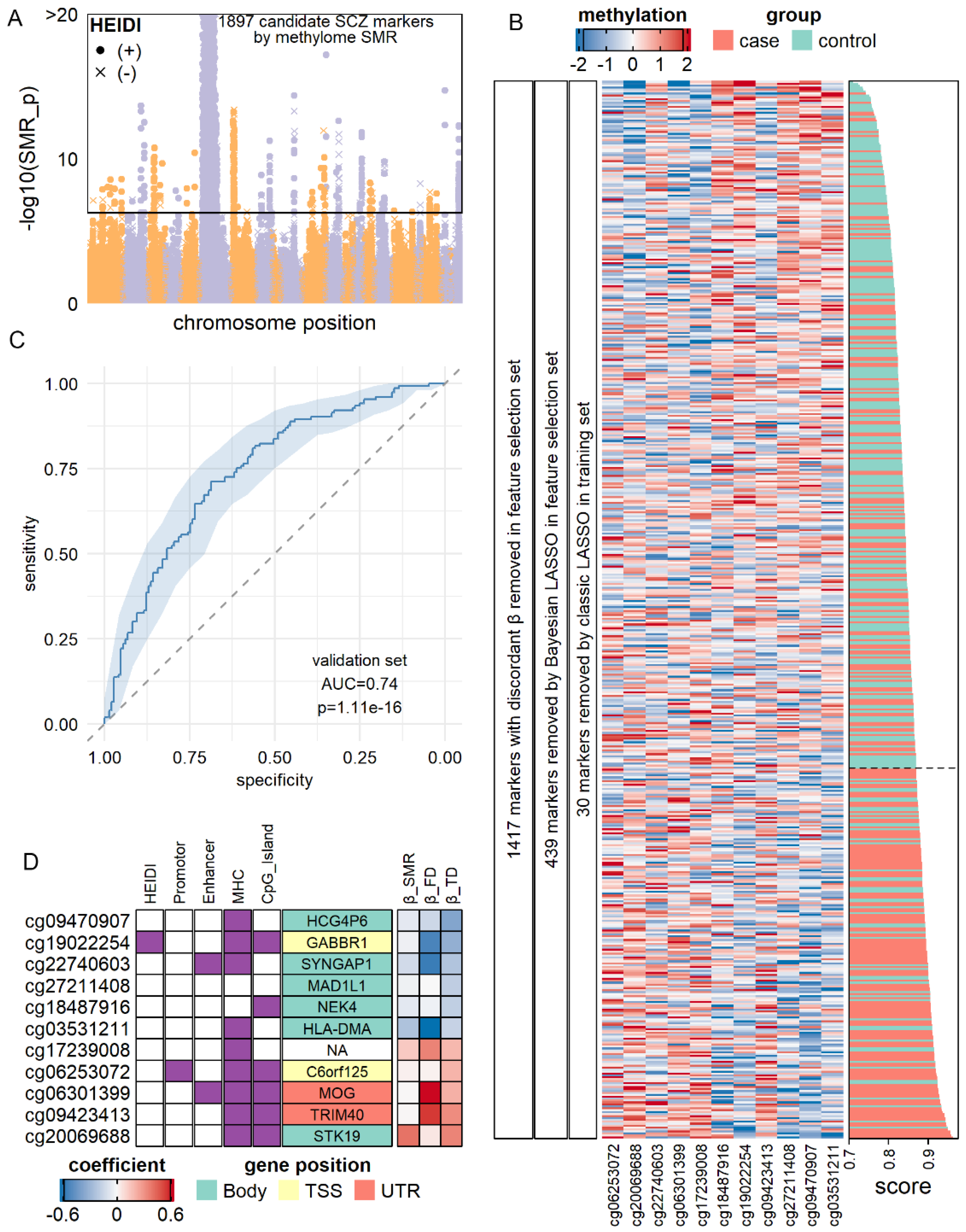
3.6. Construction of Molecular Diagnostic Models for SCZ and AD with Notable Accuracy
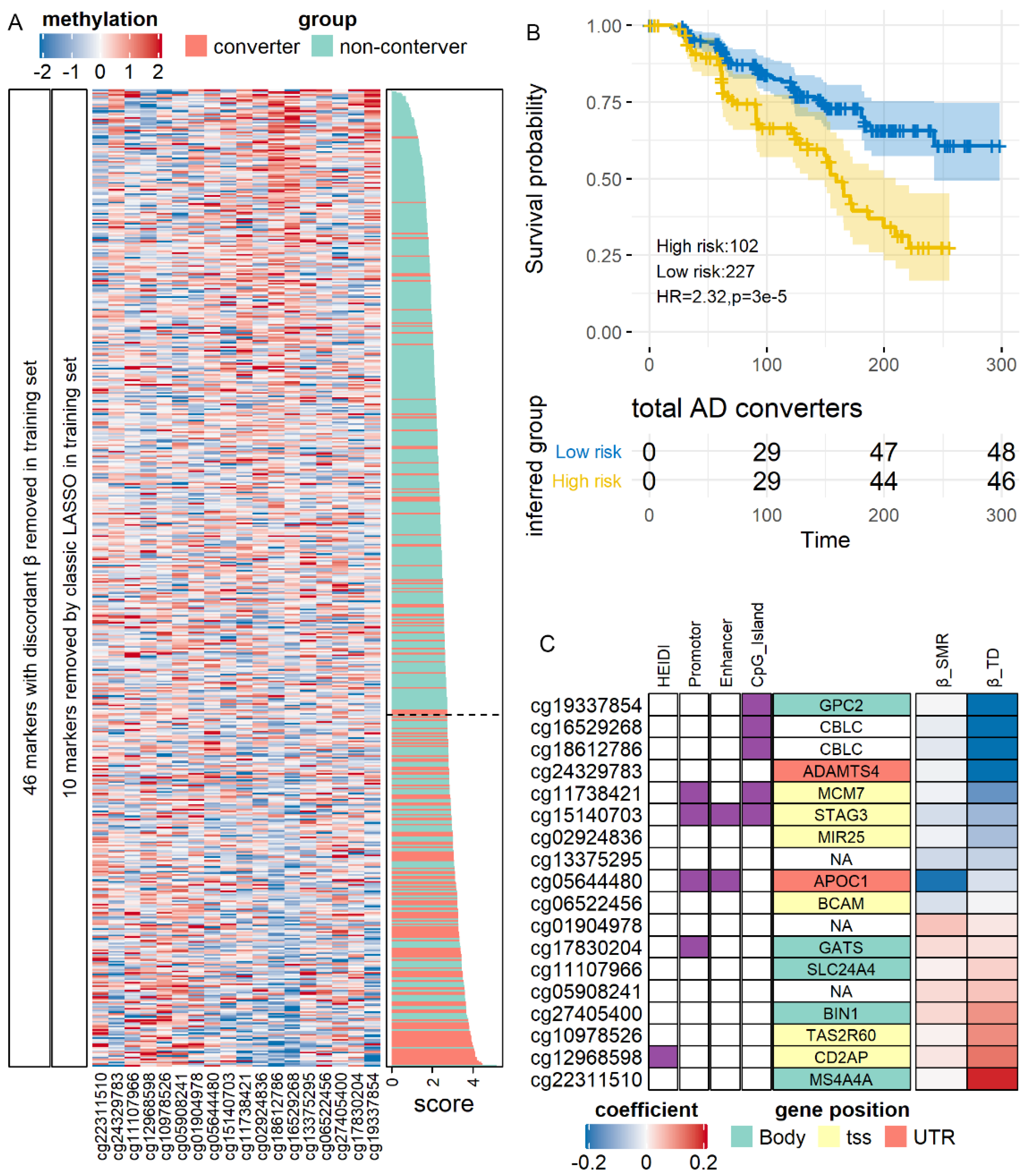
3.7. SMR-Identified Methylation Markers Predicting the Risk of AD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Insel, T.; Cuthbert, B.; Garvey, M.; Heinssen, R.; Pine, D.S.; Quinn, K.; Sanislow, C.; Wang, P. Research Domain Criteria (RDoC): Toward a new classification framework for research on mental disorders. Am. J. Psychiatry 2010, 167, 748–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, M.S.; Navarrete, F.; Sala, F.; Gasparyan, A.; Austrich-Olivares, A.; Manzanares, J. Biomarkers in Psychiatry: Concept, Definition, Types and Relevance to the Clinical Reality. Front. Psychiatry 2020, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; St. Laurent, G.; Shtokalo, D.; Antonets, D.; Vyatkin, Y.; Jones, D.; Battison, E.; Nigg, J.T. Biomarker discovery in attention deficit hyperactivity disorder: RNA sequencing of whole blood in discordant twin and case-controlled cohorts. BMC Med. Genom. 2020, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Davis, J.W.; Idler, K.; Waring, J.F.; Asque, E.; Riley-Gillis, B.; Grosskurth, S.; Srivastava, G.; Kim, S.; Kim, S.; et al. Harnessing peripheral DNA methylation differences in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) to reveal novel biomarkers of disease. Clin. Epigenet. 2020, 12, 84. [Google Scholar] [CrossRef]
- Mongan, D.; Föcking, M.; Healy, C.; Susai, S.R.; Heurich, M.; Wynne, K.; Nelson, B.; McGorry, P.D.; Amminger, G.P.; Nordentoft, M.; et al. Development of Proteomic Prediction Models for Transition to Psychotic Disorder in the Clinical High-Risk State and Psychotic Experiences in Adolescence. JAMA Psychiatry 2020, 78, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Vawter, M.P.; Iwamoto, K. Peripheral Biomarkers Revisited: Integrative Profiling of Peripheral Samples for Psychiatric Research. Biol. Psychiatry 2014, 75, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemani, G.; Bowden, J.; Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 2018, 27, R195–R208. [Google Scholar] [CrossRef] [Green Version]
- Kettunen, J.; Demirkan, A.; Würtz, P.; Draisma, H.H.M.; Haller, T.; Rawal, R.; Vaarhorst, A.; Kangas, A.J.; Lyytikäinen, L.P.; Pirinen, M.; et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun. 2016, 7, 11122. [Google Scholar] [CrossRef] [PubMed]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.P.; Raitoharju, E.; Seppälä, I.; et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Kasela, S.; et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018, 18, 447367. [Google Scholar] [CrossRef] [Green Version]
- McRae, A.F.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 2018, 8, 17605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.I.; Gaspar, H.A.; et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.J.; Yilmaz, Z.; Thornton, L.M.; Hübel, C.; Coleman, J.R.I.; Gaspar, H.A.; Bryois, J.; Hinney, A.; Leppä, V.M.; Mattheisen, M.; et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet. 2019, 51, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otowa, T.; Hek, K.; Lee, M.; Byrne, E.M.; Mirza, S.S.; Nivard, M.G.; Bigdeli, T.; Aggen, S.H.; Adkins, D.; Wolen, A.; et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol. Psychiatry 2016, 21, 1391–1399. [Google Scholar] [CrossRef]
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. Am. J. Psychiatry 2019, 176, 217–227. [Google Scholar] [CrossRef]
- Arnold, P.D.; Askland, K.D.; Barlassina, C.; Bellodi, L.; Bienvenu, O.J.; Black, D.; Bloch, M.; Brentani, H.; Burton, C.L.; Camarena, B.; et al. Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol. Psychiatry 2018, 23, 1181–1188. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.K.; Polimanti, R.; Johnson, E.C.; McClintick, J.N.; Adams, M.J.; Adkins, A.E.; Aliev, F.; Bacanu, S.A.; Batzler, A.; Bertelsen, S.; et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 2018, 21, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nievergelt, C.M.; Maihofer, A.X.; Klengel, T.; Atkinson, E.G.; Chen, C.-Y.; Choi, K.W.; Coleman, J.R.I.; Dalvie, S.; Duncan, L.E.; Gelernter, J.; et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat. Commun. 2019, 10, 4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Expanding Parkinson’s disease genetics: Novel risk loci, genomic context, causal insights and heritable risk. bioRxiv 2018, 10, 388165. [Google Scholar]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Aulchenko, Y.S.; Ripke, S.; Isaacs, A.; van Duijn, C.M. GenABEL: An R library for genome-wide association analysis. Bioinformatics 2007, 23, 1294–1296. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Skene, N.G.; Grant, S.G.N. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nho, K.; Nudelman, K.; Allen, M.; Hodges, A.; Kim, S.; Risacher, S.L.; Apostolova, L.G.; Lin, K.; Lunnon, K.; Wang, X.; et al. Genome-wide transcriptome analysis identifies novel dysregulated genes implicated in Alzheimer’s pathology. Alzheimer’s Dement. 2020, 16, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.E.; Ori, A.P.S.; Vreeker, A.; Wu, T.; Cantor, R.M.; Boks, M.P.M.; Kahn, R.S.; Olde Loohuis, L.M.; Ophoff, R.A. Whole blood transcriptome analysis in bipolar disorder reveals strong lithium effect. Psychol. Med. 2019, 50, 2575–2586. [Google Scholar] [CrossRef]
- Calligaris, R.; Banica, M.; Roncaglia, P.; Robotti, E.; Finaurini, S.; Vlachouli, C.; Antonutti, L.; Iorio, F.; Carissimo, A.; Cattaruzza, T.; et al. Blood transcriptomics of drug-naïve sporadic Parkinson’s disease patients. BMC Genom. 2015, 16, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, E.J.; Cairns, M.J.; Liu, B.; Beveridge, N.J.; Carr, V.; Kelly, B.; Scott, R.J.; Tooney, P.A. Gene expression analysis reveals schizophrenia-associated dysregulation of immune pathways in peripheral blood mononuclear cells. J. Psychiatr. Res. 2013, 47, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Pramparo, T.; Pierce, K.; Lombardo, M.V.; Carter Barnes, C.; Marinero, S.; Ahrens-Barbeau, C.; Murray, S.S.; Lopez, L.; Xu, R.; Courchesne, E. Prediction of Autism by Translation and Immune/Inflammation Coexpressed Genes in Toddlers from Pediatric Community Practices. JAMA Psychiatry 2015, 72, 386. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, K.R.; De Jong, S.; Strengman, E.; Buizer-Voskamp, J.E.; Kahn, R.S.; Boks, M.P.; Horvath, S.; Ophoff, R.A. Identification of schizophrenia-associated loci by combining DNA methylation and gene expression data from whole blood. Eur. J. Hum. Genet. 2015, 23, 1106–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leday, G.G.R.; Vértes, P.E.; Richardson, S.; Greene, J.R.; Regan, T.; Khan, S.; Henderson, R.; Freeman, T.C.; Pariante, C.M.; Harrison, N.A.; et al. Replicable and Coupled Changes in Innate and Adaptive Immune Gene Expression in Two Case-Control Studies of Blood Microarrays in Major Depressive Disorder. Biol. Psychiatry 2018, 83, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamir, R.; Klein, C.; Amar, D.; Vollstedt, E.J.; Bonin, M.; Usenovic, M.; Wong, Y.C.; Maver, A.; Poths, S.; Safer, H.; et al. Analysis of blood-based gene expression in idiopathic Parkinson disease. Neurology 2017, 89, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Sood, S.; Gallagher, I.J.; Lunnon, K.; Rullman, E.; Keohane, A.; Crossland, H.; Phillips, B.E.; Cederholm, T.; Jensen, T.; van Loon, L.J.C.; et al. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015, 16, 185. [Google Scholar] [CrossRef] [Green Version]
- Clelland, C.L.; Read, L.L.; Panek, L.J.; Nadrich, R.H.; Bancroft, C.; Clelland, J.D. Utilization of Never-Medicated Bipolar Disorder Patients towards Development and Validation of a Peripheral Biomarker Profile. PLoS ONE 2013, 8, e69082. [Google Scholar] [CrossRef] [Green Version]
- Gazestani, V.H.; Pramparo, T.; Nalabolu, S.; Kellman, B.P.; Murray, S.; Lopez, L.; Pierce, K.; Courchesne, E.; Lewis, N.E. A perturbed gene network containing PI3K–AKT, RAS–ERK and WNT–β-catenin pathways in leukocytes is linked to ASD genetics and symptom severity. Nat. Neurosci. 2019, 22, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Osborne, L.; Clive, M.; Kimmel, M.; Gispen, F.; Guintivano, J.; Brown, T.; Cox, O.; Judy, J.; Meilman, S.; Braier, A.; et al. Replication of epigenetic postpartum depression biomarkers and variation with hormone levels. Neuropsychopharmacology 2016, 41, 1648–1658. [Google Scholar] [CrossRef] [Green Version]
- Sabunciyan, S.; Maher, B.; Bahn, S.; Dickerson, F.; Yolken, R.H. Association of DNA methylation with acute mania and inflammatory markers. PLoS ONE 2015, 10, e0132001. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA methylation associates the cystine–glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratanatharathorn, A.; Boks, M.P.; Maihofer, A.X.; Aiello, A.E.; Amstadter, A.B.; Ashley-Koch, A.E.; Baker, D.G.; Beckham, J.C.; Bromet, E.; Dennis, M.; et al. Epigenome-wide association of PTSD from heterogeneous cohorts with a common multi-site analysis pipeline. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 619–630. [Google Scholar] [CrossRef]
- Chuang, Y.-H.; Paul, K.C.; Bronstein, J.M.; Bordelon, Y.; Horvath, S.; Ritz, B. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017, 9, 76. [Google Scholar] [CrossRef]
- Lohoff, F.W.; Roy, A.; Jung, J.; Longley, M.; Rosoff, D.B.; Luo, A.; O’Connell, E.; Sorcher, J.L.; Sun, H.; Schwandt, M.; et al. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: Evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol. Psychiatry 2020, 26, 2224–2237. [Google Scholar] [CrossRef] [PubMed]
- Booij, L.; Casey, K.F.; Antunes, J.M.; Szyf, M.; Joober, R.; Israël, M.; Steiger, H. DNA methylation in individuals with anorexia nervosa and in matched normal-eater controls: A genome-wide study. Int. J. Eat. Disord. 2015, 48, 874–882. [Google Scholar] [CrossRef]
- Hannon, E.; Dempster, E.; Viana, J.; Burrage, J.; Smith, A.R.; Macdonald, R.; St Clair, D.; Mustard, C.; Breen, G.; Therman, S.; et al. An integrated genetic-epigenetic analysis of schizophrenia: Evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016, 17, 176. [Google Scholar] [CrossRef] [Green Version]
- Glatting, G.; Kletting, P.; Reske, S.N.; Hohl, K.; Ring, C. Choosing the optimal fit function: Comparison of the Akaike information criterion and the F-test. Med. Phys. 2007, 34, 4285–4292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Qiao, W.; Bu, G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef] [PubMed]
- Dickens, A.M.; Sen, P.; Kempton, M.J.; Barrantes-Vidal, N.; Iyegbe, C.; Nordentoft, M.; Pollak, T.; Riecher-Rössler, A.; Ruhrmann, S.; Sachs, G.; et al. Dysregulated Lipid Metabolism Precedes Onset of Psychosis. Biol. Psychiatry 2020, 89, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Vogelzangs, N.; de Jonge, P.; Smit, J.H.; Bahn, S.; Penninx, B.W. Cytokine production capacity in depression and anxiety. Transl. Psychiatry 2016, 6, e825. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, F.P.; Borges, M.C.; Horta, B.L.; Bowden, J.; Davey Smith, G. Inflammatory Biomarkers and Risk of Schizophrenia. JAMA Psychiatry 2017, 74, 1226. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Wang, W.; Liu, Z.; Cai, W.; Yu, S.; Zhao, M.; Lin, G.N. A Comprehensive Evaluation of Cross-Omics Blood-Based Biomarkers for Neuropsychiatric Disorders. J. Pers. Med. 2021, 11, 1247. https://doi.org/10.3390/jpm11121247
Song W, Wang W, Liu Z, Cai W, Yu S, Zhao M, Lin GN. A Comprehensive Evaluation of Cross-Omics Blood-Based Biomarkers for Neuropsychiatric Disorders. Journal of Personalized Medicine. 2021; 11(12):1247. https://doi.org/10.3390/jpm11121247
Chicago/Turabian StyleSong, Weichen, Weidi Wang, Zhe Liu, Wenxiang Cai, Shunying Yu, Min Zhao, and Guan Ning Lin. 2021. "A Comprehensive Evaluation of Cross-Omics Blood-Based Biomarkers for Neuropsychiatric Disorders" Journal of Personalized Medicine 11, no. 12: 1247. https://doi.org/10.3390/jpm11121247
APA StyleSong, W., Wang, W., Liu, Z., Cai, W., Yu, S., Zhao, M., & Lin, G. N. (2021). A Comprehensive Evaluation of Cross-Omics Blood-Based Biomarkers for Neuropsychiatric Disorders. Journal of Personalized Medicine, 11(12), 1247. https://doi.org/10.3390/jpm11121247