
From DNA Copy Number Gains and Tumor Dependencies to Novel Therapeutic Targets for High-Risk Neuroblastoma

 , , , , , and
, , , , , and 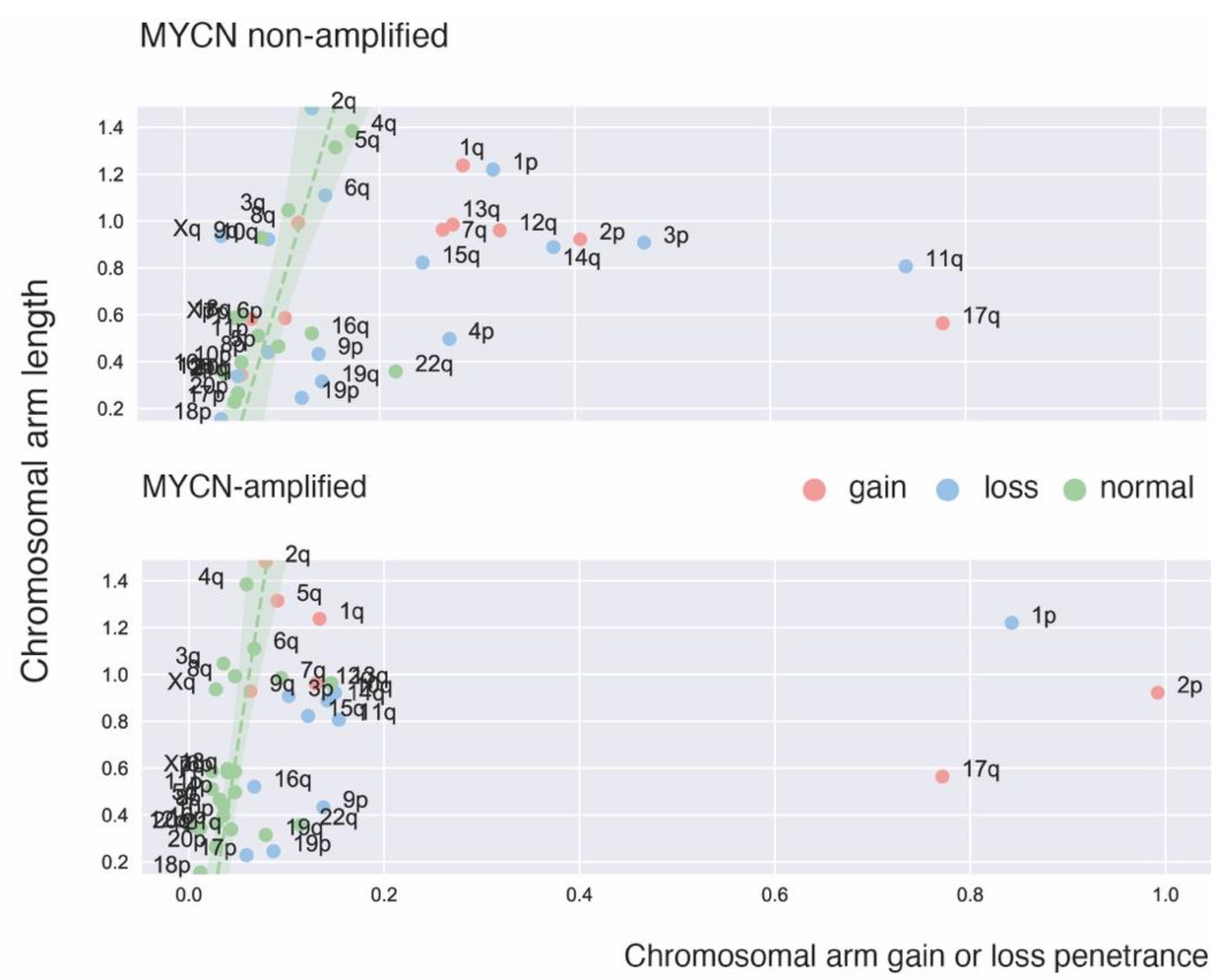{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Recurrent Focal and Large Segmental DNA Copy Number Alterations in NB
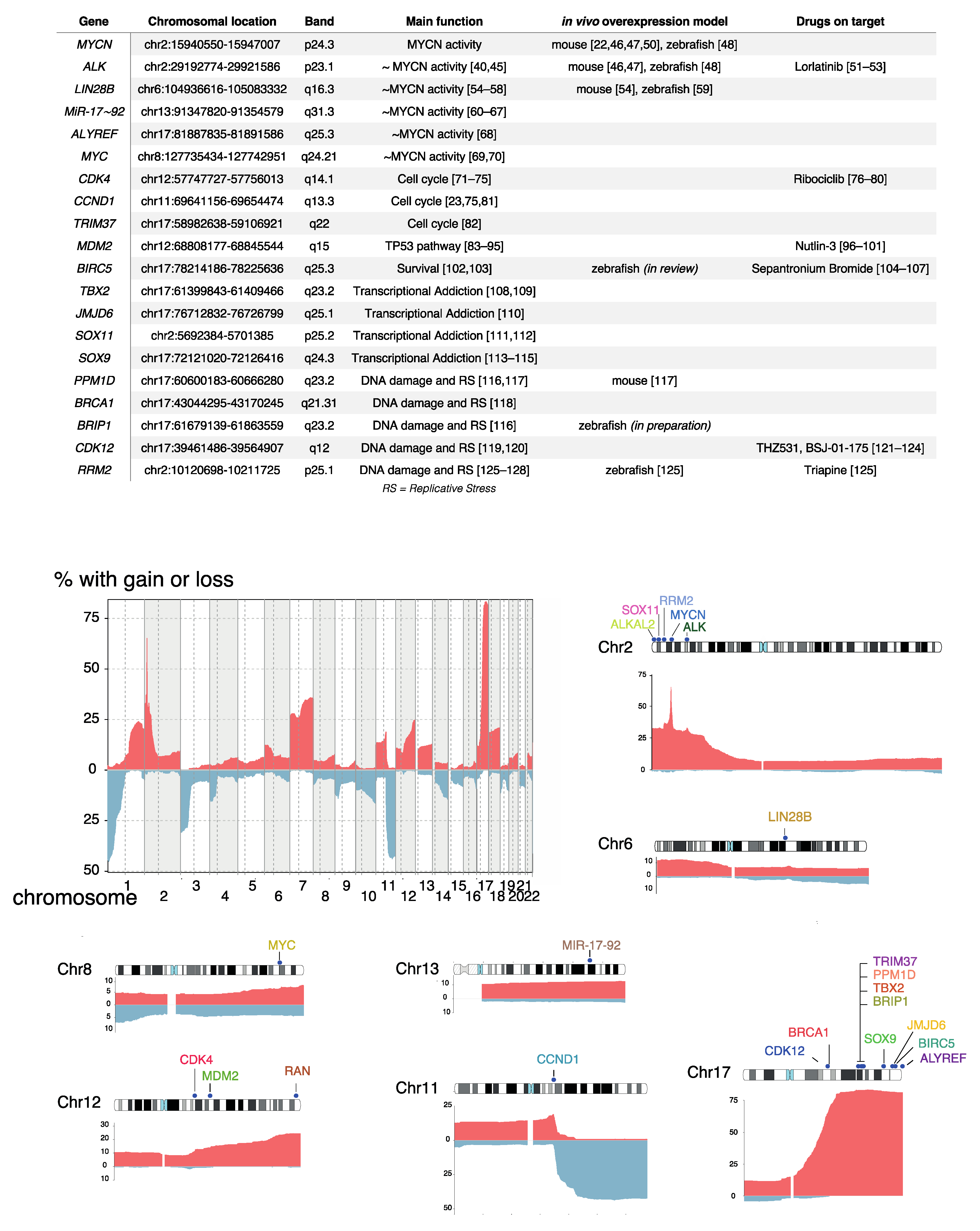
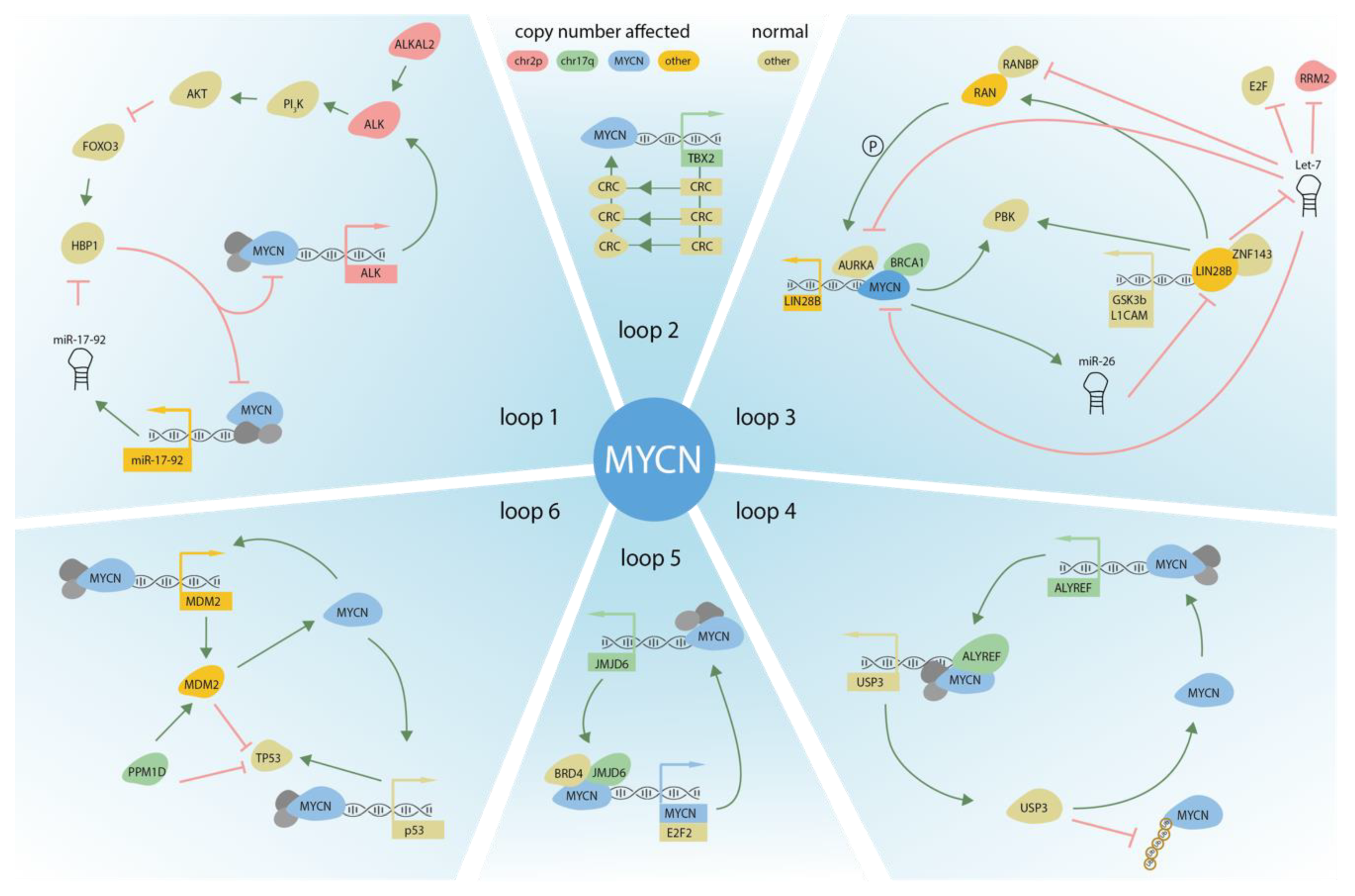
3. DNA Copy Number Affected Genes Supporting MYCN Activity
3.1. ALK

3.2. LIN28B
3.3. MiR-17~92
3.4. ALYREF
3.5. MYC

4. DNA Copy Number Affected Genes Driving Cell Cycle Activity
4.1. CDK4/CCND1
4.2. TRIM37
5. DNA Copy Number Affected Genes Involved in TP53 Pathway Control
5.1. MDM2
5.2. BIRC5
6. DNA Copy Number Affected Genes Driving Transcriptional Addiction
6.1. TBX2
6.2. JMJD6
6.3. SOX11
6.4. SOX9
7. DNA Copy Number Affected Genes Controlling DNA Damage and Replicative Stress Response
7.1. PPM1D/WIP1
7.2. BRCA1, BRIP1, CDK12
7.3. RRM2
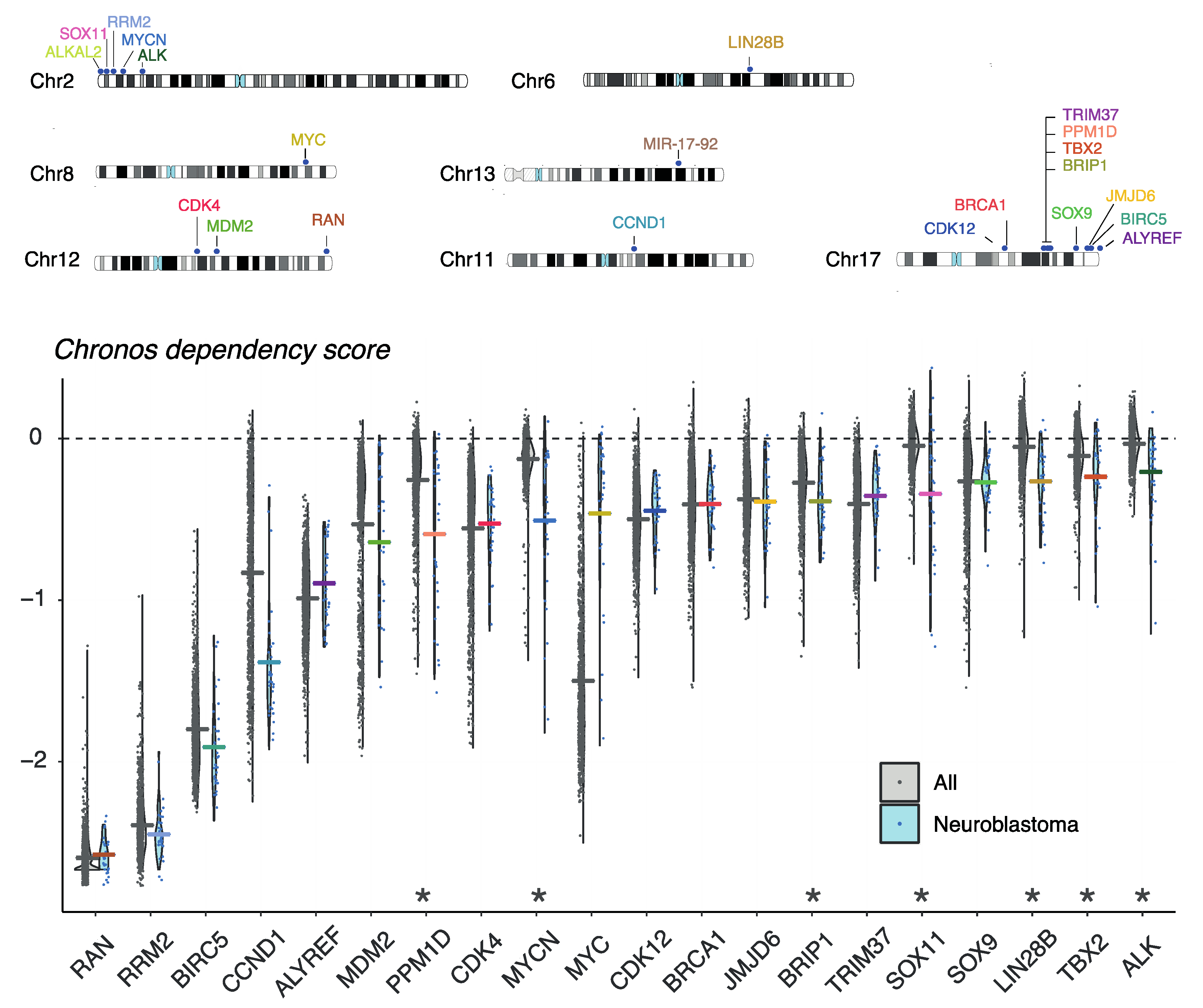
8. Evaluation of Gene Dependencies and Candidate Therapeutic Targets Using the Cancer Dependency Map
9. Current and Emerging In Vitro and In Vivo Models for Exploring the Role of DNA Copy Number Driven NB Dependencies
10. Future Perspectives to Mine CNAs for Novel Candidates and Deeper Exploration of NB Dependency Genes
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primer 2016, 2, 16078. [Google Scholar] [CrossRef]
- Schwab, M.; Varmus, H.E.; Bishop, J.M.; Grzeschik, K.H.; Naylor, S.L.; Sakaguchi, A.Y.; Brodeur, G.; Trent, J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 1984, 308, 288–291. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Ambros, P.F.; Ambros, I.M.; Strehl, S.; Bauer, S.; Luegmayr, A.; Kovar, H.; Ladenstein, R.; Fink, F.M.; Horcher, E.; Printz, G. Regression and progression in neuroblastoma. Does genetics predict tumour behaviour? Eur. J. Cancer 1995, 31, 510–515. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lequin, D.; Brugières, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther, W.; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.-K.V.; Zhang, J.; Lu, C.; Parker, M.; Bahrami, A.; Tickoo, S.K.; Heguy, A.; Pappo, A.S.; Federico, S.; Dalton, J.; et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, S.; Hiyama, E.; Onitake, Y.; Yamaoka, E.; Hiyama, K. Clinical features of ATRX or DAXX mutated neuroblastoma. J. Pediatr. Surg. 2014, 49, 1835–1838. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Schleiermacher, G.; Michels, E.; Mosseri, V.; Ribeiro, A.; Lequin, D.; Vermeulen, J.; Couturier, J.; Peuchmaur, M.; Valent, A.; et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1026–1033. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Ribeiro, A.; Klijanienko, J.; Couturier, J.; Pierron, G.; Mosseri, V.; Valent, A.; Auger, N.; Plantaz, D.; et al. Accumulation of segmental alterations determines progression in neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3122–3130. [Google Scholar] [CrossRef]
- Zeineldin, M.; Federico, S.; Chen, X.; Fan, Y.; Xu, B.; Stewart, E.; Zhou, X.; Jeon, J.; Griffiths, L.; Nguyen, R.; et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 2020, 11, 913. [Google Scholar] [CrossRef] [Green Version]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprüssel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C.; et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Green, A.A.; Hayes, F.A.; Williams, K.J.; Williams, D.L.; Tsiatis, A.A. Cytogenetic features of human neuroblastomas and cell lines. Cancer Res. 1981, 41, 4678–4686. [Google Scholar]
- Maris, J.M.; Guo, C.; Blake, D.; White, P.S.; Hogarty, M.D.; Thompson, P.M.; Rajalingam, V.; Gerbing, R.; Stram, D.O.; Matthay, K.K.; et al. Comprehensive analysis of chromosome 1p deletions in neuroblastoma. Med. Pediatr. Oncol. 2001, 36, 32–36. [Google Scholar] [CrossRef]
- White, P.S.; Thompson, P.M.; Seifried, B.A.; Sulman, E.P.; Jensen, S.J.; Guo, C.; Maris, J.M.; Hogarty, M.D.; Allen, C.; Biegel, J.A.; et al. Detailed molecular analysis of 1p36 in neuroblastoma. Med. Pediatr. Oncol. 2001, 36, 37–41. [Google Scholar] [CrossRef]
- Carén, H.; Fransson, S.; Ejeskär, K.; Kogner, P.; Martinsson, T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. Br. J. Cancer 2007, 97, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; Speleman, F.; Van Roy, N.; Laureys, G.; Brinskchmidt, C.; Christiansen, H.; Lampert, F.; Lastowska, M.; Bown, N.; Pearson, A.; et al. Multicentre analysis of patterns of DNA gains and losses in 204 neuroblastoma tumors: How many genetic subgroups are there? Med. Pediatr. Oncol. 2001, 36, 5–10. [Google Scholar] [CrossRef]
- Guo, C.; White, P.S.; Weiss, M.J.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999, 18, 4948–4957. [Google Scholar] [CrossRef] [Green Version]
- Plantaz, D.; Vandesompele, J.; Van Roy, N.; Lastowska, M.; Bown, N.; Combaret, V.; Favrot, M.C.; Delattre, O.; Michon, J.; Bénard, J.; et al. Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int. J. Cancer 2001, 91, 680–686. [Google Scholar] [CrossRef]
- Michels, E.; Vandesompele, J.; De Preter, K.; Hoebeeck, J.; Vermeulen, J.; Schramm, A.; Molenaar, J.J.; Menten, B.; Marques, B.; Stallings, R.L.; et al. ArrayCGH-based classification of neuroblastoma into genomic subgroups. Genes. Chromosomes Cancer 2007, 46, 1098–1108. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Michon, J.; Huon, I.; d’Enghien, C.D.; Klijanienko, J.; Brisse, H.; Ribeiro, A.; Mosseri, V.; Rubie, H.; Munzer, C.; et al. Chromosomal CGH identifies patients with a higher risk of relapse in neuroblastoma without MYCN amplification. Br. J. Cancer 2007, 97, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depuydt, P.; Koster, J.; Boeva, V.; Hocking, T.D.; Speleman, F.; Schleiermacher, G.; De Preter, K. Meta-mining of copy number profiles of high-risk neuroblastoma tumors. Sci. Data 2018, 5, 180240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roy, N.; Cheng, N.C.; Laureys, G.; Opdenakker, G.; Versteeg, R.; Speleman, F. Molecular cytogenetic analysis of 1;17 translocations in neuroblastoma. Eur. J. Cancer 1995, 31A, 530–535. [Google Scholar] [CrossRef]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Lastowska, M.; Van Roy, N.; Bown, N.; Speleman, F.; Lunec, J.; Strachan, T.; Pearson, A.D.; Jackson, M.S. Molecular cytogenetic delineation of 17q translocation breakpoints in neuroblastoma cell lines. Genes. Chromosomes Cancer 1998, 23, 116–122. [Google Scholar] [CrossRef]
- Vandesompele, J.; Michels, E.; De Preter, K.; Menten, B.; Schramm, A.; Eggert, A.; Ambros, P.F.; Combaret, V.; Francotte, N.; Antonacci, F.; et al. Identification of 2 putative critical segments of 17q gain in neuroblastoma through integrative genomics. Int. J. Cancer 2008, 122, 1177–1182. [Google Scholar] [CrossRef]
- Depuydt, P.; Boeva, V.; Hocking, T.D.; Cannoodt, R.; Ambros, I.M.; Ambros, P.F.; Asgharzadeh, S.; Attiyeh, E.F.; Combaret, V.; Defferrari, R.; et al. Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J. Natl. Cancer Inst. 2018, 110, 1084–1093. [Google Scholar] [CrossRef]
- Vandesompele, J.; Baudis, M.; De Preter, K.; Van Roy, N.; Ambros, P.; Bown, N.; Brinkschmidt, C.; Christiansen, H.; Combaret, V.; Lastowska, M.; et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2280–2299. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Helmsauer, K.; Valieva, M.E.; Ali, S.; Chamorro González, R.; Schöpflin, R.; Röefzaad, C.; Bei, Y.; Dorado Garcia, H.; Rodriguez-Fos, E.; Puiggròs, M.; et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 2020, 11, 5823. [Google Scholar] [CrossRef] [PubMed]
- Koche, R.P.; Rodriguez-Fos, E.; Helmsauer, K.; Burkert, M.; MacArthur, I.C.; Maag, J.; Chamorro, R.; Munoz-Perez, N.; Puiggròs, M.; Dorado Garcia, H.; et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 2020, 52, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Kumps, C.; Fieuw, A.; Mestdagh, P.; Menten, B.; Lefever, S.; Pattyn, F.; De Brouwer, S.; Sante, T.; Schulte, J.H.; Schramm, A.; et al. Focal DNA copy number changes in neuroblastoma target MYCN regulated genes. PLoS ONE 2013, 8, e52321. [Google Scholar]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra91. [Google Scholar] [CrossRef]
- Zhu, S.; Lee, J.-S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Unno, K.; Chalmers, Z.R.; Pamarthy, S.; Vatapalli, R.; Rodriguez, Y.; Lysy, B.; Mok, H.; Sagar, V.; Han, H.; Yoo, Y.A.; et al. Activated ALK Cooperates with N-Myc via Wnt/β-Catenin Signaling to Induce Neuroendocrine Prostate Cancer. Cancer Res. 2021, 81, 2157–2170. [Google Scholar] [CrossRef]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Kayser, K.; Groshen, S.G.; Chioda, M.; Thurm, H.C.; Chen, J.; Peltz, G.; Granger, M.; Maris, J.; Matthay, K.K.; et al. Phase I trial of lorlatinib in patients with ALK-driven refractory or relapsed neuroblastoma: A New Approaches to Neuroblastoma Consortium study. J. Clin. Oncol. 2020, 38, 10504. [Google Scholar] [CrossRef]
- Phase 1 Study of Lorlatinib (PF-06463922), an Oral Small Molecule Inhibitor of ALK/ROS1, for Patients with ALK-Driven Relapsed or Refractory Neuroblastoma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03107988 (accessed on 22 November 2021).
- Real World Data Collection among Pediatric Neuroblastoma Patients Treated with Lorlatinib through Expanded Access Program. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04753658?term=lorlatinib&cond=Neuroblastoma&draw=2&rank=1 (accessed on 22 November 2021).
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, A.; Van Peer, G.; Carter, D.R.; Gartlgruber, M.; Herrmann, C.; Agarwal, S.; Helsmoortel, H.H.; Althoff, K.; Molenaar, J.J.; Cheung, B.B.; et al. MYCN-driven regulatory mechanisms controlling LIN28B in neuroblastoma. Cancer Lett. 2015, 366, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Cox, J.; Annam, J.; Weingart, M.; Essien, G.; Rathi, K.S.; Rokita, J.L.; Khurana, P.; Cuya, S.M.; Bosse, K.R.; et al. LIN28B promotes neuroblastoma metastasis and regulates PDZ binding kinase. Neoplasia 2020, 22, 231–241. [Google Scholar] [CrossRef]
- Schnepp, R.W.; Khurana, P.; Attiyeh, E.F.; Raman, P.; Chodosh, S.E.; Oldridge, D.A.; Gagliardi, M.E.; Conkrite, K.L.; Asgharzadeh, S.; Seeger, R.C.; et al. A LIN28B-RAN-AURKA Signaling Network Promotes Neuroblastoma Tumorigenesis. Cancer Cell 2015, 28, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, T.; Shi, H.; Mariani, L.; Abraham, B.J.; Durbin, A.D.; Zimmerman, M.W.; Powers, J.T.; Missios, P.; Ross, K.N.; Perez-Atayde, A.R.; et al. LIN28B regulates transcription and potentiates MYCN-induced neuroblastoma through binding to ZNF143 at target gene promotors. Proc. Natl. Acad. Sci. USA 2020, 117, 16516–16526. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-alpha-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Fiori, M.E.; Albini, S.; Cifaldi, L.; Giovinazzi, S.; Forloni, M.; Boldrini, R.; Donfrancesco, A.; Federici, V.; Giacomini, P.; et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS ONE 2008, 3, e2236. [Google Scholar] [CrossRef] [Green Version]
- Dews, M.; Fox, J.L.; Hultine, S.; Sundaram, P.; Wang, W.; Liu, Y.Y.; Furth, E.; Enders, G.H.; El-Deiry, W.; Schelter, J.M.; et al. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010, 70, 8233–8246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestdagh, P.; Boström, A.-K.; Impens, F.; Fredlund, E.; Van Peer, G.; De Antonellis, P.; von Stedingk, K.; Ghesquière, B.; Schulte, S.; Dews, M.; et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol. Cell 2010, 40, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.-C.; Vidigal, J.A.; Mu, P.; Yao, E.; Singh, I.; González, A.J.; Concepcion, C.P.; Bonetti, C.; Ogrodowski, P.; Carver, B.; et al. An allelic series of miR-17∼92–mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nat. Genet. 2015, 47, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Fredlund, E.; Pattyn, F.; Schulte, J.H.; Muth, D.; Vermeulen, J.; Kumps, C.; Schlierf, S.; De Preter, K.; Van Roy, N.; et al. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene 2010, 29, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.; Seneviratne, J.A.; Kanikevich, M.; Chang, W.; Mayoh, C.; Venkat, P.; Du, Y.; Jiang, C.; Salib, A.; Koach, J.; et al. An ALYREF-MYCN coactivator complex drives neuroblastoma tumorigenesis through effects on USP3 and MYCN stability. Nat. Commun. 2021, 12, 1881. [Google Scholar] [CrossRef]
- Van Roy, N.; Vandesompele, J.; Menten, B.; Nilsson, H.; De Smet, E.; Rocchi, M.; De Paepe, A.; Påhlman, S.; Speleman, F. Translocation-excision-deletion-amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes. Chromosomes Cancer 2006, 45, 107–117. [Google Scholar] [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef] [Green Version]
- Van Roy, N.; Forus, A.; Myklebost, O.; Cheng, N.C.; Versteeg, R.; Speleman, F. Identification of two distinct chromosome 12-derived amplification units in neuroblastoma cell line NGP. Cancer Genet. Cytogenet. 1995, 82, 151–154. [Google Scholar]
- Kranenburg, O.; Scharnhorst, V.; Van der Eb, A.J.; Zantema, A. Inhibition of cyclin-dependent kinase activity triggers neuronal differentiation of mouse neuroblastoma cells. J. Cell Biol. 1995, 131, 227–234. [Google Scholar] [CrossRef]
- Gogolin, S.; Ehemann, V.; Becker, G.; Brueckner, L.M.; Dreidax, D.; Bannert, S.; Nolte, I.; Savelyeva, L.; Bell, E.; Westermann, F. CDK4 inhibition restores G1-S arrest in MYCN-amplified neuroblastoma cells in the context of doxorubicin-induced DNA damage. Cell Cycle 2013, 12, 1091–1104. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Caron, H.; Geoerger, B.; Eggert, A.; Schleiermacher, G.; Brock, P.; Valteau-Couanet, D.; Chesler, L.; Schulte, J.H.; De Preter, K.; et al. Accelerating drug development for neuroblastoma-New Drug Development Strategy: An Innovative Therapies for Children with Cancer, European Network for Cancer Research in Children and Adolescents and International Society of Paediatric Oncology Europe Neuroblastoma project. Expert Opin. Drug Discov. 2017, 12, 801–811. [Google Scholar] [PubMed]
- Rihani, A.; Vandesompele, J.; Speleman, F.; Van Maerken, T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer Cell Int. 2015, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Schubert, N.A.; Schild, L.; van Oirschot, S.; Keller, K.M.; Alles, L.K.; Vernooij, L.; Nulle, M.E.; Dolman, M.E.M.; van den Boogaard, M.L.; Molenaar, J.J. Combined targeting of the p53 and pRb pathway in neuroblastoma does not lead to synergistic responses. Eur. J. Cancer 2021, 142, 1–9. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Ebus, M.E.; Koster, J.; van Sluis, P.; van Noesel, C.J.M.; Versteeg, R.; Caron, H.N. Cyclin D1 and CDK4 Activity Contribute to the Undifferentiated Phenotype in Neuroblastoma. Cancer Res. 2008, 68, 2599–2609. [Google Scholar] [CrossRef] [Green Version]
- Meitinger, F.; Ohta, M.; Lee, K.-Y.; Watanabe, S.; Davis, R.L.; Anzola, J.V.; Kabeche, R.; Jenkins, D.A.; Shiau, A.K.; Desai, A.; et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020, 585, 440–446. [Google Scholar] [CrossRef]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar] [PubMed]
- Corvi, R.; Savelyeva, L.; Breit, S.; Wenzel, A.; Handgretinger, R.; Barak, J.; Oren, M.; Amler, L.; Schwab, M. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 1995, 10, 1081–1086. [Google Scholar] [PubMed]
- Corvi, R.; Savelyeva, L.; Amler, L.; Handgretinger, R.; Schwab, M. Cytogenetic evolution of MYCN and MDM2 amplification in the neuroblastoma LS tumour and its cell line. Eur. J. Cancer 1995, 31A, 520–523. [Google Scholar] [CrossRef]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.-K.V.; Boos, J.; et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clin. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lin, Y.; Barbieri, E.; Burlingame, S.; Hicks, J.; Ludwig, A.; Shohet, J.M. Mdm2 deficiency suppresses MYCN-Driven neuroblastoma tumorigenesis in vivo. Neoplasia 2009, 11, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Van Maerken, T.; Vandesompele, J.; Rihani, A.; De Paepe, A.; Speleman, F. Escape from p53-mediated tumor surveillance in neuroblastoma: Switching off the p14ARF-MDM2-p53 axis. Cell Death Differ. 2009, 16, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Carr, J.; Bell, E.; Pearson, A.D.J.; Kees, U.R.; Beris, H.; Lunec, J.; Tweddle, D.A. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006, 66, 2138–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreidax, D.; Gogolin, S.; Schroeder, C.; Muth, D.; Brueckner, L.M.; Hess, E.M.; Zapatka, M.; Theißen, J.; Fischer, M.; Ehemann, V.; et al. Low p14ARF expression in neuroblastoma cells is associated with repressed histone mark status, and enforced expression induces growth arrest and apoptosis. Hum. Mol. Genet. 2013, 22, 1735–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Slack, A.D.; Chen, Z.; Ludwig, A.D.; Hicks, J.; Shohet, J.M. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007, 67, 2448–2455. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Gu, L.; Zhang, H.; Zhou, M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle 2011, 10, 2994–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhang, H.; He, J.; Li, J.; Huang, M.; Zhou, M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 2012, 31, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Milazzo, G.; Rajapakshe, K.; Bernardi, R.; Chen, Z.; Barbieri, E.; Koster, J.; Perini, G.; Coarfa, C.; Shohet, J.M. MYCN acts as a direct co-regulator of p53 in MYCN amplified neuroblastoma. Oncotarget 2018, 9, 20323–20338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippé, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, E.; Mehta, P.; Chen, Z.; Zhang, L.; Slack, A.; Berg, S.; Shohet, J.M. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol. Cancer Ther. 2006, 5, 2358–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Rothweiler, F.; Klassert, D.; von Deimling, A.; Weber, K.; Fehse, B.; Kammerer, B.; Doerr, H.W.; Cinatl, J. Reversal of P-glycoprotein-mediated multidrug resistance by the murine double minute 2 antagonist nutlin-3. Cancer Res. 2009, 69, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, X.; Rajaei, M.; Youn, J.Y.; Zafar, A.; Deokar, H.; Buolamwini, J.K.; Yang, J.; Foster, J.H.; Zhou, J.; et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers 2020, 12, 3651. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. 2021, 496, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Adida, C.; Berrebi, D.; Peuchmaur, M.; Reyes-Mugica, M.; Altieri, D.C. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet 1998, 351, 882–883. [Google Scholar] [CrossRef]
- Islam, A.; Kageyama, H.; Takada, N.; Kawamoto, T.; Takayasu, H.; Isogai, E.; Ohira, M.; Hashizume, K.; Kobayashi, H.; Kaneko, Y.; et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 2000, 19, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef] [Green Version]
- Hipp, N.I.; Christner, L.; Wirth, T.; Mueller-Klieser, W.; Walenta, S.; Schröck, E.; Debatin, K.-M.; Beltinger, C. MYCN and survivin cooperatively contribute to malignant transformation of fibroblasts. Carcinogenesis 2014, 35, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Voges, Y.; Rothweiler, F.; Weipert, F.; Zia-Ahmad, A.; Cinatl, J.; von Deimling, A.; Westermann, F.; Rödel, F.; Wass, M.N.; et al. Testing of the Survivin Suppressant YM155 in a Large Panel of Drug-Resistant Neuroblastoma Cell Lines. Cancers 2020, 12, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholizadeh, S.; Dolman, E.M.; Wieriks, R.; Sparidans, R.W.; Hennink, W.E.; Kok, R.J. Anti-GD2 Immunoliposomes for Targeted Delivery of the Survivin Inhibitor Sepantronium Bromide (YM155) to Neuroblastoma Tumor Cells. Pharm. Res. 2018, 35, 85. [Google Scholar] [CrossRef] [Green Version]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Decaesteker, B.; Denecker, G.; Van Neste, C.; Dolman, E.M.; Van Loocke, W.; Gartlgruber, M.; Nunes, C.; De Vloed, F.; Depuydt, P.; Verboom, K.; et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat. Commun. 2018, 9, 4866. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 3319. [Google Scholar] [CrossRef] [Green Version]
- Decaesteker, B.; Louwagie, A.; Loontiens, S.; Vloed, F.D.; Roels, J.; Vanhauwaert, S.; Brouwer, S.D.; Sanders, E.; Denecker, G.; D’haene, E.; et al. SOX11 Is a Lineage-Dependency Factor and Master Epigenetic Regulator in Neuroblastoma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Afanasyeva, E.A.; Gartlgruber, M.; Ryl, T.; Decaesteker, B.; Denecker, G.; Mönke, G.; Toprak, U.H.; Florez, A.; Torkov, A.; Dreidax, D.; et al. Kalirin-RAC controls nucleokinetic migration in ADRN-type neuroblastoma. Life Sci. Alliance 2021, 4, e201900332. [Google Scholar] [CrossRef]
- Mondal, T.; Juvvuna, P.K.; Kirkeby, A.; Mitra, S.; Kosalai, S.T.; Traxler, L.; Hertwig, F.; Wernig-Zorc, S.; Miranda, C.; Deland, L.; et al. Sense-Antisense lncRNA Pair Encoded by Locus 6p22.3 Determines Neuroblastoma Susceptibility via the USP36-CHD7-SOX9 Regulatory Axis. Cancer Cell 2018, 33, 417–434.e7. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Hannum, D.F.; Zhai, Y.; Hill, S.F.; Ricardo, D.; Lou, W.; Skidmore, J.M.; Sanchez, G.; Saiakhova, A.; Bielas, S.L.; et al. CHD7 promotes neural progenitor differentiation in embryonic stem cells via altered chromatin accessibility and nascent gene expression. Sci. Rep. 2020, 10, 17445. [Google Scholar] [CrossRef]
- Yang, C.-L.; Serra-Roma, A.; Gualandi, M.; Bodmer, N.; Niggli, F.; Schulte, J.H.; Bode, P.K.; Shakhova, O. Lineage-restricted sympathoadrenal progenitors confer neuroblastoma origin and its tumorigenicity. Oncotarget 2020, 11, 2357–2371. [Google Scholar] [CrossRef] [PubMed]
- Saito-Ohara, F.; Imoto, I.; Inoue, J.; Hosoi, H.; Nakagawara, A.; Sugimoto, T.; Inazawa, J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003, 63, 1876–1883. [Google Scholar] [PubMed]
- Milosevic, J.; Fransson, S.; Gulyas, M.; Gallo-Oller, G.; Olsen, T.K.; Treis, D.; Wickström, M.; Elfman, L.H.; Sveinbjornsson, B.; Hertwig, F.; et al. PPM1D Is a Neuroblastoma Oncogene and Therapeutic Target in Childhood Neural Tumors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.G.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Jiang, J.; Kaltheuner, I.H.; Iniguez, A.B.; Anand, K.; Ferguson, F.M.; Ficarro, S.B.; Seong, B.K.A.; Greifenberg, A.K.; Dust, S.; et al. Structure-activity relationship study of THZ531 derivatives enables the discovery of BSJ-01-175 as a dual CDK12/13 covalent inhibitor with efficacy in Ewing sarcoma. Eur. J. Med. Chem. 2021, 221, 113481. [Google Scholar] [CrossRef]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef]
- Nunes, C.; Depestel, L.; Mus, L.; Keller, K.; Delhaye, L.; Louwagie, A.; Rishfi, M.; Dolman, E.; Olexiouk, V.; Bartenhagen, C.; et al. RRM2 Is a Target for Synthetic Lethal Interactions with Replication Stress Checkpoint Addiction in High-Risk Neuroblastoma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mazzu, Y.Z.; Armenia, J.; Chakraborty, G.; Yoshikawa, Y.; Coggins, S.A.; Nandakumar, S.; Gerke, T.A.; Pomerantz, M.M.; Qiu, X.; Zhao, H.; et al. A Novel Mechanism Driving Poor-Prognosis Prostate Cancer: Overexpression of the DNA Repair Gene, Ribonucleotide Reductase Small Subunit M2 (RRM2). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4480–4492. [Google Scholar] [CrossRef] [Green Version]
- Fatkhutdinov, N.; Sproesser, K.; Krepler, C.; Liu, Q.; Brafford, P.A.; Herlyn, M.; Aird, K.M.; Zhang, R. Targeting RRM2 and Mutant BRAF Is a Novel Combinatorial Strategy for Melanoma. Mol. Cancer Res. MCR 2016, 14, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Ohmura, S.; Marchetto, A.; Orth, M.F.; Li, J.; Jabar, S.; Ranft, A.; Vinca, E.; Ceranski, K.; Carreño-Gonzalez, M.J.; Romero-Pérez, L.; et al. Translational evidence for RRM2 as a prognostic biomarker and therapeutic target in Ewing sarcoma. Mol. Cancer 2021, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, I.; Kumps, C.; Claeys, S.; Lindner, S.; Beckers, A.; Janssens, E.; Carter, D.R.; Cazes, A.; Cheung, B.B.; De Mariano, M.; et al. Upregulation of MAPK Negative Feedback Regulators and RET in Mutant ALK Neuroblastoma: Implications for Targeted Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3327–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mus, L.M.; Lambertz, I.; Claeys, S.; Kumps, C.; Van Loocke, W.; Van Neste, C.; Umapathy, G.; Vaapil, M.; Bartenhagen, C.; Laureys, G.; et al. The ETS transcription factor ETV5 is a target of activated ALK in neuroblastoma contributing to increased tumour aggressiveness. Sci. Rep. 2020, 10, 218. [Google Scholar] [CrossRef]
- Claeys, S.; Denecker, G.; Durinck, K.; Decaesteker, B.; Mus, L.M.; Loontiens, S.; Vanhauwaert, S.; Althoff, K.; Wigerup, C.; Bexell, D.; et al. ALK positively regulates MYCN activity through repression of HBP1 expression. Oncogene 2019, 38, 2690–2705. [Google Scholar] [CrossRef]
- Emdal, K.B.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Lundby, A.; Claeys, S.; De Preter, K.; Speleman, F.; Francavilla, C.; Olsen, J.V. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci. Signal. 2018, 11, eaap9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoya-Reina, O.C.; Li, W.; Arceo, M.; Plescher, M.; Bullova, P.; Pui, H.; Kaucka, M.; Kharchenko, P.; Martinsson, T.; Holmberg, J.; et al. Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat. Commun. 2021, 12, 5309. [Google Scholar] [CrossRef] [PubMed]
- Borenäs, M.; Umapathy, G.; Lai, W.-Y.; Lind, D.E.; Witek, B.; Guan, J.; Mendoza-Garcia, P.; Masudi, T.; Claeys, A.; Chuang, T.-P.; et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021, 40, e105784. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Koster, J.; Ebus, M.E.; van Sluis, P.; Westerhout, E.M.; de Preter, K.; Gisselsson, D.; Øra, I.; Speleman, F.; Caron, H.N.; et al. Copy number defects of G1-Cell cycle genes in neuroblastoma are frequent and correlate with high expression of E2F target genes and a poor prognosis. Genes. Chromosomes Cancer 2012, 51, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Waber, P.G.; Chen, J.; Nisen, P.D. Infrequency of MDM2 gene amplification in pediatric solid tumors and lack of association with p53 mutations in adult squamous cell carcinomas. Cancer Res. 1993, 53, 6028–6030. [Google Scholar] [PubMed]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Velculescu, V.E.; Madden, S.L.; Zhang, L.; Lash, A.E.; Yu, J.; Rago, C.; Lal, A.; Wang, C.J.; Beaudry, G.A.; Ciriello, K.M.; et al. Analysis of human transcriptomes. Nat. Genet. 1999, 23, 387–388. [Google Scholar] [CrossRef]
- Qi, J.; Dong, Z.; Liu, J.; Peery, R.C.; Zhang, S.; Liu, J.-Y.; Zhang, J.-T. Effective Targeting of the Survivin Dimerization Interface with Small-Molecule Inhibitors. Cancer Res. 2016, 76, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Berezov, A.; Cai, Z.; Freudenberg, J.A.; Zhang, H.; Cheng, X.; Thompson, T.; Murali, R.; Greene, M.I.; Wang, Q. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene 2012, 31, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Fenstermaker, R.A.; Figel, S.A.; Qiu, J.; Barone, T.A.; Dharma, S.S.; Winograd, E.K.; Galbo, P.M.; Wiltsie, L.M.; Ciesielski, M.J. Survivin Monoclonal Antibodies Detect Survivin Cell Surface Expression and Inhibit Tumor Growth In Vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2642–2652. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, L.; Tang, C.; Yin, C. Co-Delivery of Doxorubicin and Survivin shRNA-Expressing Plasmid Via Microenvironment-Responsive Dendritic Mesoporous Silica Nanoparticles for Synergistic Cancer Therapy. Pharm. Res. 2017, 34, 2829–2841. [Google Scholar] [CrossRef]
- Nucleosome-Bound SOX2 and SOX11 Structures Elucidate Pioneer Factor Function|Nature. Available online: https://www.nature.com/articles/s41586-020-2195-y (accessed on 4 October 2021).
- Richter, M.; Dayaram, T.; Gilmartin, A.G.; Ganji, G.; Pemmasani, S.K.; Van Der Key, H.; Shohet, J.M.; Donehower, L.A.; Kumar, R. WIP1 Phosphatase as a Potential Therapeutic Target in Neuroblastoma. PLoS ONE 2015, 10, e0115635. [Google Scholar] [CrossRef] [PubMed]
- De Wyn, J.; Zimmerman, M.W.; Weichert-Leahey, N.; Nunes, C.; Cheung, B.B.; Abraham, B.J.; Beckers, A.; Volders, P.-J.; Decaesteker, B.; Carter, D.R.; et al. MEIS2 Is an Adrenergic Core Regulatory Transcription Factor Involved in Early Initiation of TH-MYCN-Driven Neuroblastoma Formation. Cancers 2021, 13, 4783. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, R.A.; Peng, Y.; Norris, Z.A.; Tholey, R.M.; Talbott, V.A.; Liang, Q.; Ai, Y.; Miller, K.; Lal, S.; Cozzitorto, J.A.; et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol. Cancer Res. MCR 2013, 11, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DepMap Achilles 18Q3 Public. Figshare. Available online: https://figshare.com/articles/dataset/DepMap_Achilles_18Q3_public/6931364/1 (accessed on 3 August 2018).
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A CRISPR Cell Population Dynamics Model. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A First-Generation Pediatric Cancer Dependency Map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, X.; Weichert-Leahey, N.; Dong, Z.; Zhang, C.; Lopez, G.; Tao, T.; He, S.; Wood, A.C.; Oldridge, D.; et al. LMO1 Synergizes with MYCN to Promote Neuroblastoma Initiation and Metastasis. Cancer Cell 2017, 32, 310–323.e5. [Google Scholar] [CrossRef] [Green Version]
- Loontiens, S.; Depestel, L.; Vanhauwaert, S.; Dewyn, G.; Gistelinck, C.; Verboom, K.; Van Loocke, W.; Matthijssens, F.; Willaert, A.; Vandesompele, J.; et al. Purification of high-quality RNA from a small number of fluorescence activated cell sorted zebrafish cells for RNA sequencing purposes. BMC Genom. 2019, 20, 228. [Google Scholar] [CrossRef]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.-D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef] [Green Version]
- García-López, J.; Wallace, K.; Otero, J.H.; Olsen, R.; Wang, Y.-D.; Finkelstein, D.; Gudenas, B.L.; Rehg, J.E.; Northcott, P.; Davidoff, A.M.; et al. Large 1p36 Deletions Affecting Arid1a Locus Facilitate Mycn-Driven Oncogenesis in Neuroblastoma. Cell Rep. 2020, 30, 454–464.e5. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.A.; Zhang, S.; Sengupta, S.; Ma, H.; Bell, G.W.; Horton, B.; Sharma, B.; George, R.E.; Spranger, S.; Jaenisch, R. Formation of Human Neuroblastoma in Mouse-Human Neural Crest Chimeras. Cell Stem Cell 2020, 26, 579–592.e6. [Google Scholar] [CrossRef] [PubMed]
- Mus, L.M.; Van Haver, S.; Popovic, M.; Trypsteen, W.; Lefever, S.; Zeltner, N.; Ogando, Y.; Jacobs, E.Z.; Denecker, G.; Sanders, E.; et al. Recurrent chromosomal imbalances provide selective advantage to human embryonic stem cells under enhanced replicative stress conditions. Genes. Chromosomes Cancer 2021, 60, 272–281. [Google Scholar] [CrossRef]
- Akavia, U.D.; Litvin, O.; Kim, J.; Sanchez-Garcia, F.; Kotliar, D.; Causton, H.C.; Pochanard, P.; Mozes, E.; Garraway, L.A.; Pe’er, D. An integrated approach to uncover drivers of cancer. Cell 2010, 143, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, E.; Calzone, L.; Michoel, T. Integrative Multi-omics Module Network Inference with Lemon-Tree. PLoS Comput. Biol. 2015, 11, e1003983. [Google Scholar] [CrossRef] [Green Version]
- Champion, M.; Brennan, K.; Croonenborghs, T.; Gentles, A.J.; Pochet, N.; Gevaert, O. Module Analysis Captures Pancancer Genetically and Epigenetically Deregulated Cancer Driver Genes for Smoking and Antiviral Response. EBioMedicine 2018, 27, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Bense, R.D.; Urzúa-Traslaviña, C.G.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. Transcriptional effects of copy number alterations in a large set of human cancers. Nat. Commun. 2020, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Zhang, C.; Qu, L.; Huang, J.; Jiang, L.; Liu, J.; Pinello, L.; Yuan, G.-C.; Shou, C. Gene regulatory pattern analysis reveals essential role of core transcriptional factors’ activation in triple-negative breast cancer. Oncotarget 2017, 8, 21938–21953. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Radvanyi, F.; Elati, M. CoRegNet: Reconstruction and integrated analysis of co-regulatory networks. Bioinformatics 2015, 31, 3066–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, E.O.; Aytes, A.; Jones, S.J.; Subramaniam, P.S.; Giorgi, F.M.; Douglass, E.F.; Tagore, S.; Chu, B.; Vasciaveo, A.; Zheng, S.; et al. A modular master regulator landscape controls cancer transcriptional identity. Cell 2021, 184, 334–351.e20. [Google Scholar] [CrossRef]
- Silverbush, D.; Cristea, S.; Yanovich-Arad, G.; Geiger, T.; Beerenwinkel, N.; Sharan, R. Simultaneous Integration of Multi-omics Data Improves the Identification of Cancer Driver Modules. Cell Syst. 2019, 8, 456–466.e5. [Google Scholar] [CrossRef]
- Colaprico, A.; Olsen, C.; Bailey, M.H.; Odom, G.J.; Terkelsen, T.; Silva, T.C.; Olsen, A.V.; Cantini, L.; Zinovyev, A.; Barillot, E.; et al. Interpreting pathways to discover cancer driver genes with Moonlight. Nat. Commun. 2020, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Rajbhandari, P.; Lopez, G.; Capdevila, C.; Salvatori, B.; Yu, J.; Rodriguez-Barrueco, R.; Martinez, D.; Yarmarkovich, M.; Weichert-Leahey, N.; Abraham, B.J.; et al. Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer Discov. 2018, 8, 582–599. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, Y.; Wei, Z.; Chang, X. Coexpression network analysis identifies transcriptional modules associated with genomic alterations in neuroblastoma. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2018, 1864, 2341–2348. [Google Scholar] [CrossRef]
- Zhang, L.; Lv, C.; Jin, Y.; Cheng, G.; Fu, Y.; Yuan, D.; Tao, Y.; Guo, Y.; Ni, X.; Shi, T. Deep Learning-Based Multi-Omics Data Integration Reveals Two Prognostic Subtypes in High-Risk Neuroblastoma. Front. Genet. 2018, 9, 477. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Candido-Ferreira, I.; Repapi, E.; Gavriouchkina, D.; Senanayake, U.; Ling, I.T.C.; Telenius, J.; Taylor, S.; Hughes, J.; Sauka-Spengler, T. Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Dev. Cell 2019, 51, 255–276.e7. [Google Scholar] [CrossRef] [Green Version]
- Leo, L.; Colonna Romano, N. Emerging Single-Cell Technological Approaches to Investigate Chromatin Dynamics and Centromere Regulation in Human Health and Disease. Int. J. Mol. Sci. 2021, 22, 8809. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Camphausen, K.; Shankavaram, U. Pan-Cancer Analysis of Potential Synthetic Lethal Drug Targets Specific to Alterations in DNA Damage Response. Front. Oncol. 2019, 9, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decaesteker, B.; Durinck, K.; Van Roy, N.; De Wilde, B.; Van Neste, C.; Van Haver, S.; Roberts, S.; De Preter, K.; Vermeirssen, V.; Speleman, F. From DNA Copy Number Gains and Tumor Dependencies to Novel Therapeutic Targets for High-Risk Neuroblastoma. J. Pers. Med. 2021, 11, 1286. https://doi.org/10.3390/jpm11121286
Decaesteker B, Durinck K, Van Roy N, De Wilde B, Van Neste C, Van Haver S, Roberts S, De Preter K, Vermeirssen V, Speleman F. From DNA Copy Number Gains and Tumor Dependencies to Novel Therapeutic Targets for High-Risk Neuroblastoma. Journal of Personalized Medicine. 2021; 11(12):1286. https://doi.org/10.3390/jpm11121286
Chicago/Turabian StyleDecaesteker, Bieke, Kaat Durinck, Nadine Van Roy, Bram De Wilde, Christophe Van Neste, Stéphane Van Haver, Stephen Roberts, Katleen De Preter, Vanessa Vermeirssen, and Frank Speleman. 2021. "From DNA Copy Number Gains and Tumor Dependencies to Novel Therapeutic Targets for High-Risk Neuroblastoma" Journal of Personalized Medicine 11, no. 12: 1286. https://doi.org/10.3390/jpm11121286
APA StyleDecaesteker, B., Durinck, K., Van Roy, N., De Wilde, B., Van Neste, C., Van Haver, S., Roberts, S., De Preter, K., Vermeirssen, V., & Speleman, F. (2021). From DNA Copy Number Gains and Tumor Dependencies to Novel Therapeutic Targets for High-Risk Neuroblastoma. Journal of Personalized Medicine, 11(12), 1286. https://doi.org/10.3390/jpm11121286