
Kinesin Family Member C1 (KIFC1/HSET): A Potential Actionable Biomarker of Early Stage Breast Tumorigenesis and Progression of High-Risk Lesions

 , ,
, , {kind=link}
Abstract
:1. Introduction
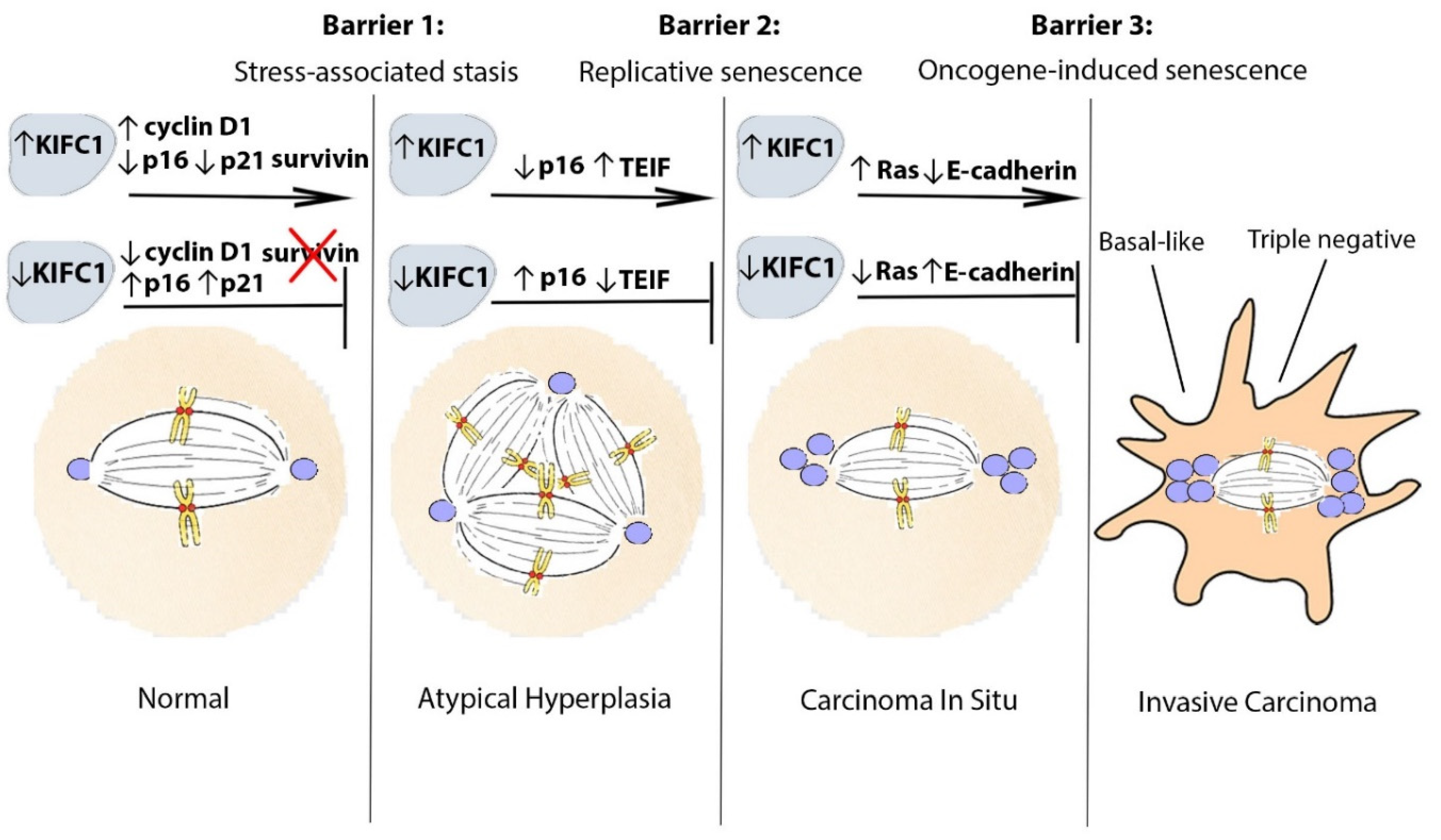
2. From the Beginning: Transition of Pre-Neoplastic Cells to an Oncogenic Phenotype
3. Bypassing the Stress-Associated Stasis Barrier: KIFC1 and Cell Cycle Deregulation
4. Bypassing the Replicative Cellular Senescence Barrier: KIFC1 and Loss of Telomere Function and Genomic Stability
5. Bypassing the Oncogene-Induced Senescence Barrier: KIFC1 and Ras Signaling
6. Luminal or Basal-like: KIFC1 and Intrinsic Subtype Specification
7. Future of Breast Cancer Risk Management: Evaluating and Targeting KIFC1 in High-Risk Patient Subpopulations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, P.A.; Goodwin, P.J. Breast Cancer Survivorship: Where Are We Today? Adv. Exp. Med. Biol. 2015, 862, 1–8. [Google Scholar]
- Troester, M.A.; Sun, X.; Allott, E.H.; Geradts, J.; Cohen, S.M.; Tse, C.K.; Kirk, E.L.; Thorne, L.B.; Mathews, M.; Li, Y.; et al. Racial Differences in PAM50 Subtypes in the Carolina Breast Cancer Study. J. Natl. Cancer Inst. 2018, 110, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, D.; Hu, H.; Rhie, S.K.; Gamazon, E.R.; Cherniack, A.D.; Liu, J.; Yoshimatsu, T.F.; Pitt, J.J.; Hoadley, K.A.; Troester, M.; et al. Comparison of Breast Cancer Molecular Features and Survival by African and European Ancestry in The Cancer Genome Atlas. JAMA Oncol. 2017, 3, 1654–1662. [Google Scholar] [CrossRef]
- Russo, J.; Calaf, G.; Sohi, N.; Tahin, Q.; Zhang, P.L.; Alvarado, M.E.; Estrada, S.; Russo, I.H. Critical Steps in Breast Carcinogenesis. Ann. N. Y. Acad. Sci. 1993, 698, 1–20. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. The Pathway of Neoplastic Transformation of Human Breast Epithelial Cells. Radiat. Res. 2001, 155, 151–154. [Google Scholar] [CrossRef]
- Russo, J.; Yang, X.; Hu, Y.F.; Bove, B.A.; Huang, Y.; Silva, I.D.; Tahin, Q.; Wu, Y.; Higgy, N.; Zekri, A.; et al. Biological and Molecular Basis of Human Breast Cancer. Front. Biosci. 1998, 3, D944–D960. [Google Scholar] [CrossRef]
- Russo, J.; Calaf, G.; Russo, I.H. A Critical Approach to the Malignant Transformation of Human Breast Epithelial Cells with Chemical Carcinogens. Crit. Rev. Oncog. 1993, 4, 403–417. [Google Scholar]
- Page, D.L.; Dupont, W.D. Anatomic Markers of Human Premalignancy and Risk of Breast Cancer. Cancer 1990, 66, 1326–1335. [Google Scholar] [CrossRef]
- Garbe, J.C.; Bhattacharya, S.; Merchant, B.; Bassett, E.; Swisshelm, K.; Feiler, H.S.; Wyrobek, A.J.; Stampfer, M.R. Molecular Distinctions between Stasis and Telomere Attrition Senescence Barriers Shown by Long-Term Culture of Normal Human Mammary Epithelial Cells. Cancer Res. 2009, 69, 7557–7568. [Google Scholar] [CrossRef] [Green Version]
- Garbe, J.C.; Holst, C.R.; Bassett, E.; Tlsty, T.; Stampfer, M.R. Inactivation of p53 Function in Cultured Human Mammary Epithelial Cells Turns the Telomere-Length Dependent Senescence Barrier from Agonescence into Crisis. Cell Cycle 2007, 6, 1927–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, J.C.; Vrba, L.; Sputova, K.; Fuchs, L.; Novak, P.; Brothman, A.R.; Jackson, M.; Chin, K.; LaBarge, M.A.; Watts, G.; et al. Immortalization of Normal Human Mammary Epithelial Cells in Two Steps by Direct Targeting of Senescence Barriers Does not Require Gross Genomic Alterations. Cell Cycle 2014, 13, 3423–3435. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Weaver, L.N.; Ems-McClung, S.C.; Walczak, C.E. Kinesin-14 Family Proteins HSET/XCTK2 Control Spindle Length by Cross-Linking and Sliding Microtubules. Mol. Biol. Cell 2009, 20, 1348–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.Y. A Clinical Overview of Centrosome Amplification in Human Cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef]
- D’Assoro, A.B.; Lingle, W.L.; Salisbury, J.L. Centrosome Amplification and the Development of Cancer. Oncogene 2002, 21, 6146–6153. [Google Scholar] [CrossRef] [Green Version]
- Lingle, W.L.; Barrett, S.L.; Negron, V.C.; D’Assoro, A.B.; Boeneman, K.; Liu, W.; Whitehead, C.M.; Reynolds, C.; Salisbury, J.L. Centrosome Amplification Drives Chromosomal Instability in Breast Tumor Development. Proc. Natl. Acad. Sci. USA 2002, 99, 1978–1983. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.A.M.; Mesquita, M.; Cunha, A.I.; Cardoso, J.; Carapeta, S.; Laranjeira, C.; Pinto, A.E.; Pereira-Leal, J.B.; Dias-Pereira, A.; Bettencourt-Dias, M.; et al. Centrosome Amplification Arises before Neoplasia and Increases Upon p53 Loss in Tumorigenesis. J. Cell Biol. 2018, 217, 2353–2363. [Google Scholar] [CrossRef]
- Sridharan, D.M.; Enerio, S.; LaBarge, M.A.; Stampfer, M.M.; Pluth, J.M. Lesion Complexity Drives Age Related Cancer Susceptibility in Human Mammary Epithelial Cells. Aging (Albany NY) 2017, 9, 665–686. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Pyo, J.H.; Na, H.J.; Jeon, H.J.; Kim, Y.S.; Arking, R.; Yoo, M.A. Increased Centrosome Amplification in Aged Stem Cells of the Drosophila Midgut. Biochem. Biophys Res. Commun. 2014, 450, 961–965. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Liu, T.; Tse, G.M.; Sun, B.; Niu, R.; Li, H.M.; Wang, H.; Yang, Y.; Ye, X.; Wang, Y.; et al. Increased Expression of Centrosomal Alpha, Gamma-Tubulin in Atypical Ductal Hyperplasia and Carcinoma of the Breast. Cancer Sci. 2009, 100, 580–587. [Google Scholar] [CrossRef]
- Pannu, V.; Mittal, K.; Cantuaria, G.; Reid, M.D.; Li, X.; Donthamsetty, S.; McBride, M.; Klimov, S.; Osan, R.; Gupta, M.V.; et al. Rampant Centrosome Amplification Underlies more Aggressive Disease Course of Triple Negative Breast Cancers. Oncotarget 2015, 6, 10487–10497. [Google Scholar] [CrossRef] [Green Version]
- Morgunova, V.; Kordyukova, M.; Mikhaleva, E.A.; Butenko, I.; Pobeguts, O.V.; Kalmykova, A. Loss of Telomere Silencing Is Accompanied by Dysfunction of Polo Kinase and Centrosomes during Drosophila Oogenesis and Early Development. PLoS ONE 2021, 16, e0258156. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Frias, C.; Genesca, A.; Tusell, L. Progressive Telomere Dysfunction Causes Cytokinesis Failure and Leads to the Accumulation of Polyploid Cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Sun, Y.; McNutt, M.A.; Sun, Q.; Hou, L.; Liu, H.; Shen, Q.; Ling, Y.; Chi, Y.; Zhang, B. Localization of TEIF in the Centrosome and its Functional Association with Centrosome Amplification in DNA Damage, Telomere Dysfunction and Human Cancers. Oncogene 2009, 28, 1549–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome Dysfunction: A Link between Senescence and Tumor Immunity. Signal Transduct. Target. Ther. 2020, 5, 107. [Google Scholar] [CrossRef]
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome Amplification Can Initiate Tumorigenesis in Flies. Cell 2008, 133, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Godinho, S.A.; Picone, R.; Burute, M.; Dagher, R.; Su, Y.; Leung, C.T.; Polyak, K.; Brugge, J.S.; Thery, M.; Pellman, D. Oncogene-like Induction of Cellular Invasion from Centrosome Amplification. Nature 2014, 510, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5. [Google Scholar] [CrossRef] [Green Version]
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome Amplification Induces High Grade Features and Is Prognostic of Worse Outcomes in Breast Cancer. BMC Cancer 2016, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle Multipolarity Is Prevented by Centrosomal Clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, Y.; Zheng, B.; Zou, Y.; Wang, C.; Wu, P.; Wang, J.; Chen, H.; Du, P.; Liang, B.; et al. KIFC1, a Novel Potential Prognostic Factor and Therapeutic Target in Hepatocellular Carcinoma. Int. J. Oncol. 2018, 52, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhan, P.; Zhou, Z.; Xing, Z.; Zhu, S.; Ma, C.; Li, Q.; Zhu, Q.; Miao, Y.; Zhang, J.; et al. The Overexpression of KIFC1 Was Associated with the Proliferation and Prognosis of Non-Small Cell Lung Cancer. J. Thorac. Dis. 2016, 8, 2911–2923. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, Y.; Lang, Z.; Huang, J.; Zou, Z. Prognostic and Clinicopathological Significance of Kinesin Family Member C1 in Various Cancers: A Meta-Analysis. Medicine (Baltimore) 2019, 98, e17346. [Google Scholar] [CrossRef] [PubMed]
- Kostecka, L.G.; Olseen, A.; Kang, K.; Torga, G.; Pienta, K.J.; Amend, S.R. High KIFC1 Expression Is Associated with Poor Prognosis in Prostate Cancer. Med. Oncol. 2021, 38, 47. [Google Scholar] [CrossRef]
- Pannu, V.; Rida, P.C.; Ogden, A.; Turaga, R.C.; Donthamsetty, S.; Bowen, N.J.; Rudd, K.; Gupta, M.V.; Reid, M.D.; Cantuaria, G.; et al. HSET Overexpression Fuels Tumor Progression Via Centrosome Clustering-Independent Mechanisms in Breast Cancer Patients. Oncotarget 2015, 6, 6076–6091. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, W.; Chen, D.; Boohaker, R.J.; Zhai, L.; Padmalayam, I.; Wennerberg, K.; Xu, B.; Zhang, W. KIFC1 Is a Novel Potential Therapeutic Target for Breast Cancer. Cancer Biol. Ther. 2015, 16, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.X.; Yang, W.X. KIFC1: A Promising Chemotherapy Target for Cancer Treatment? Oncotarget 2016, 7, 48656–48670. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.L.; Ham, R.G.; Stampfer, M.R. Serum-Free Growth of Human Mammary Epithelial Cells: Rapid Clonal Growth in Defined Medium and Extended Serial Passage with Pituitary Extract. Proc. Natl. Acad. Sci. USA 1984, 81, 5435–5439. [Google Scholar] [CrossRef] [Green Version]
- Band, V. Preneoplastic Transformation of Human Mammary Epithelial Cells. Semin. Cancer Biol. 1995, 6, 185–192. [Google Scholar] [CrossRef]
- Derventzi, A.; Rattan, S.I.; Gonos, E.S. Molecular Links between Cellular Mortality and Immortality (Review). Anticancer Res. 1996, 16, 2901–2910. [Google Scholar]
- Olsen, C.L.; Gardie, B.; Yaswen, P.; Stampfer, M.R. Raf-1-Induced Growth Arrest in Human Mammary Epithelial Cells Is p16-Independent and Is Overcome in Immortal Cells during Conversion. Oncogene 2002, 21, 6328–6339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfer, M.R.; LaBarge, M.A.; Garbe, J.C. An Integrated Human Mammary Epithelial Cell Culture System for Studying Carcinogenesis and Aging. In Cell and Molecular Biology of Breast Cancer; Humana Press: Totowa, NJ, USA, 2013; pp. 323–361. [Google Scholar]
- Lee, J.K.; Garbe, J.C.; Vrba, L.; Miyano, M.; Futscher, B.W.; Stampfer, M.R.; LaBarge, M.A. Age and the Means of by Passing Stasis Influence the Intrinsic Subtype of Immortalized Human Mammary Epithelial Cells. Front. Cell Dev. Biol. 2015, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 Tumor Suppressor Gene in Cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Romanov, S.R.; Kozakiewicz, B.K.; Holst, C.R.; Stampfer, M.R.; Haupt, L.M.; Tlsty, T.D. Normal Human Mammary Epithelial Cells Spontaneously Escape Senescence and Acquire Genomic Changes. Nature 2001, 409, 633–637. [Google Scholar] [CrossRef]
- Stampfer, M.R.; Bodnar, A.; Garbe, J.; Wong, M.; Pan, A.; Villeponteau, B.; Yaswen, P. Gradual Phenotypic Conversion Associated with Immortalization of Cultured Human Mammary Epithelial Cells. Mol. Biol. Cell 1997, 8, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Hopfer, U.; Jacobberger, J.W.; Gruenert, D.C.; Eckert, R.L.; Jat, P.S.; Whitsett, J.A. Immortalization of Epithelial Cells. Am. J. Physiol. 1996, 270, C1–C11. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E.; Werbin, H. Toward a Molecular Understanding of Human Breast Cancer: A Hypothesis. Breast Cancer Res. Treat. 1993, 25, 83–94. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Meng, L.; Stampfer, M.; Gabai, V.L.; Yaglom, J.A. Oncogenes Induce Senescence with Incomplete Growth Arrest and Suppress the DNA Damage Response in Immortalized Cells. Aging Cell 2011, 10, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human Breast Cancer Cells Generated by Oncogenic Transformation of Primary Mammary Epithelial Cells. Genes. Dev. 2001, 15, 50–65. [Google Scholar] [CrossRef] [Green Version]
- Khodjakov, A.; Rieder, C.L. Centrosomes Enhance the Fidelity of Cytokinesis in Vertebrates and Are Required for Cell Cycle Progression. J. Cell Biol. 2001, 153, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Piel, M.; Nordberg, J.; Euteneuer, U.; Bornens, M. Centrosome-Dependent Exit of Cytokinesis in Animal Cells. Science 2001, 291, 1550–1553. [Google Scholar] [CrossRef]
- Zyss, D.; Gergely, F. Centrosome Function in Cancer: Guilty or Innocent? Trends Cell Biol. 2009, 19, 334–346. [Google Scholar] [CrossRef]
- Sabat-Pospiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting Centrosome Amplification, an Achilles’ Heel of Cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a Tragedy: Mitotic Catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Lansky, Z.; Bajer, S.; Fink, G.; Kasprzak, A.A.; Diez, S. The Human Kinesin-14 HSET Tracks the Tips of Growing Microtubules in vitro. Cytoskeleton 2013, 70, 515–521. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Pollinger, B.; Ohmer, M.; Hofmann, F.; Nigg, E.A.; Hemmings, B.A.; Wartmann, M. Acentrosomal Spindle Organization Renders Cancer Cells Dependent on the Kinesin HSET. J. Cell Sci. 2012, 125, 5391–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar Spindle Pole Coalescence Is a Major Source of Kinetochore Mis-Attachment and Chromosome Mis-Segregation in Cancer Cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.S.; Holland, A.J. The Impact of Mitotic Errors on Cell Proliferation and Tumorigenesis. Genes. Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jusino, S.; Fernandez-Padin, F.M.; Saavedra, H.I. Centrosome Aberrations and Chromosome Instability Contribute to Tumorigenesis and Intra-Tumor Heterogeneity. J. Cancer Metastasis. Treat. 2018, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- McDermott, K.M.; Zhang, J.; Holst, C.R.; Kozakiewicz, B.K.; Singla, V.; Tlsty, T.D. p16(INK4a) Prevents Centrosome Dysfunction and Genomic Instability in Primary Cells. PLoS Biol. 2006, 4, e51. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.; Feijoo, P.; Bernal, A.; Ercilla, A.; Agell, N.; Genesca, A.; Tusell, L. Centrosome Aberrations in Human Mammary Epithelial Cells Driven by Cooperative Interactions between p16INK4a Deficiency and Telomere-Dependent Genotoxic Stress. Oncotarget 2015, 6, 28238–28256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns, T. Centrosome Duplication. a Centriolar Pas De Deux. Cell 2001, 105, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Giacinti, C.; Giordano, A. RB and Cell Cycle Progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Iovino, F.; Lentini, L.; Amato, A.; Di Leonardo, A. RB Acute Loss Induces Centrosome Amplification and Aneuploidy in Murine Primary Fibroblasts. Mol. Cancer 2006, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Vande Woude, G.F. Abnormal Centrosome Amplification in the Absence of p53. Science 1996, 271, 1744–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markey, M.P.; Bergseid, J.; Bosco, E.E.; Stengel, K.; Xu, H.; Mayhew, C.N.; Schwemberger, S.J.; Braden, W.A.; Jiang, Y.; Babcock, G.F.; et al. Loss of the Retinoblastoma Tumor Suppressor: Differential Action on Transcriptional Programs Related to Cell Cycle Control and Immune Function. Oncogene 2007, 26, 6307–6318. [Google Scholar] [CrossRef] [Green Version]
- Mittal, K.; Wei, G.; Kaur, J.; Choi, D.H.; Reid, M.D.; Rida, P.C.G.; Aneja, R. KIFC1 as a Novel Therapeutic Target for p53 Mutant Colorectal Cancer. J. Clin. Oncol. 2018, 36, e15585. [Google Scholar] [CrossRef]
- Kim, N.; Song, K. KIFC1 Is Essential for Bipolar Spindle Formation and Genomic Stability in the Primary Human Fibroblast IMR-90 Cell. Cell Struct. Funct. 2013, 38, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A Gene Expression Signature of Retinoblastoma Loss-of-Function Is a Predictive Biomarker of Resistance to Palbociclib in Breast Cancer Cell Lines and Is Prognostic in Patients with ER Positive Early Breast Cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef] [Green Version]
- Tusell, L.; Pampalona, J.; Soler, D.; Frias, C.; Genesca, A. Different Outcomes of Telomere-Dependent Anaphase Bridges. Biochem. Soc. Trans. 2010, 38, 1698–1703. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Abe, S.; Huda, N.; Tu, L.; Beam, M.J.; Grimes, B.; Gilley, D. Telomere Fusions in Early Human Breast Carcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, B. Expression of TEIF Protein in Colorectal Tumors and Its Correlation with Centrosome Abnormality. Ai Zheng 2009, 28, 1277–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, G.; Sun, L.; Meng, L.; Hu, C.; Wang, X.; Shi, Z.; Hu, C.; Han, Y.; Yang, Q.; Cao, L.; et al. The ATM and ATR Kinases Regulate Centrosome Clustering and Tumor Recurrence by Targeting KIFC1 Phosphorylation. Nat. Commun. 2021, 12, 20. [Google Scholar] [CrossRef]
- Zeng, X.; Shaikh, F.Y.; Harrison, M.K.; Adon, A.M.; Trimboli, A.J.; Carroll, K.A.; Sharma, N.; Timmers, C.; Chodosh, L.A.; Leone, G.; et al. The Ras Oncogene Signals Centrosome Amplification in Mammary Epithelial Cells through Cyclin D1/Cdk4 and Nek2. Oncogene 2010, 29, 5103–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Rhys, A.D.; Monteiro, P.; Smith, C.; Vaghela, M.; Arnandis, T.; Kato, T.; Leitinger, B.; Sahai, E.; McAinsh, A.; Charras, G.; et al. Loss of E-Cadherin Provides Tolerance to Centrosome Amplification in Epithelial Cancer Cells. J. Cell Biol. 2018, 217, 195–209. [Google Scholar] [CrossRef]
- Li, J.; Diao, H.; Guan, X.; Tian, X. Kinesin Family Member C1 (KIFC1) Regulated by Centrosome Protein E (CENPE) Promotes Proliferation, Migration, and Epithelial-Mesenchymal Transition of Ovarian Cancer. Med. Sci. Monit. 2020, 26, e927869. [Google Scholar] [CrossRef]
- Patel, S.; Zambruni, J.P.; Palazuelos, D.; Legesse, H.; Ndiaye, N.F.; Detjen, A.; Aboubaker, S. Rethinking the Scale up of Integrated Management of Childhood Illness. BMJ 2018, 362, k2993. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.; Rida, P.C.; Aneja, R. Prognostic value of CA20, a Score Based on Centrosome Amplification-Associated Genes, in Breast Tumors. Sci. Rep. 2017, 7, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Weekes, D.; Drosopoulos, K.; Gazinska, P.; Noel, E.; Rashid, M.; Mirza, H.; Quist, J.; Braso-Maristany, F.; Mathew, S.; et al. Integrated Genomics and Functional Validation Identifies Malignant Cell Specific Dependencies in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 1044. [Google Scholar] [CrossRef]
- Ogden, A.; Rida, P.C.G.; Aneja, R. Centrosome amplification: A Suspect in Breast Cancer and Racial Disparities. Endocr. Relat. Cancer 2017, 24, T47–T64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, A.; Garlapati, C.; Li, X.B.; Turaga, R.C.; Oprea-Ilies, G.; Wright, N.; Bhattarai, S.; Mittal, K.; Wetherilt, C.S.; Krishnamurti, U.; et al. Multi-Institutional Study of Nuclear KIFC1 as a Biomarker of Poor Prognosis in African American Women with Triple-Negative Breast Cancer. Sci. Rep. 2017, 7, 42289. [Google Scholar] [CrossRef] [Green Version]
- Bryc, K.; Auton, A.; Nelson, M.R.; Oksenberg, J.R.; Hauser, S.L.; Williams, S.; Froment, A.; Bodo, J.M.; Wambebe, C.; Tishkoff, S.A.; et al. Genome-Wide Patterns of Population Structure and Admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. USA 2010, 107, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooker, S.E., Jr.; Woods-Burnham, L.; Bathina, M.; Lloyd, S.; Gorjala, P.; Mitra, R.; Nonn, L.; Kimbro, K.S.; Kittles, R.A. Genetic Ancestry Analysis Reveals Misclassification of Commonly Used Cancer Cell Lines. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, D.J.; Walls, A.L. Atypical Breast Hyperplasia; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- van Seijen, M.; Lips, E.H.; Thompson, A.M.; Nik-Zainal, S.; Futreal, A.; Hwang, E.S.; Verschuur, E.; Lane, J.; Jonkers, J.; Rea, D.W.; et al. Ductal Carcinoma in Situ: To Treat or Not to Treat, That Is the Question. Br. J. Cancer 2019, 121, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to Suppress Multipolar Divisions in Cancer Cells with Extra Centrosomes. Genes. Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Mikule, K.; Wang, W.; Su, N.; Petteruti, P.; Gharahdaghi, F.; Code, E.; Zhu, X.; Jacques, K.; Lai, Z.; et al. Discovery and Mechanistic Study of a Small Molecule Inhibitor for Motor Protein KIFC1. ACS Chem. Biol. 2013, 8, 2201–2208. [Google Scholar] [CrossRef]
- Marthiens, V.; Rujano, M.A.; Pennetier, C.; Tessier, S.; Paul-Gilloteaux, P.; Basto, R. Centrosome Amplification Causes Microcephaly. Nat. Cell Biol. 2013, 15, 731–740. [Google Scholar] [CrossRef]
- Vitre, B.; Holland, A.J.; Kulukian, A.; Shoshani, O.; Hirai, M.; Wang, Y.; Maldonado, M.; Cho, T.; Boubaker, J.; Swing, D.A.; et al. Chronic Centrosome Amplification without Tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E6321–E6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, N.; Gong, Z.; Kittles, R.; Natarajan, R.; Jovanovic-Talisman, T.; Rida, P.; LaBarge, M.; Seewaldt, V. Kinesin Family Member C1 (KIFC1/HSET): A Potential Actionable Biomarker of Early Stage Breast Tumorigenesis and Progression of High-Risk Lesions. J. Pers. Med. 2021, 11, 1361. https://doi.org/10.3390/jpm11121361
Wright N, Gong Z, Kittles R, Natarajan R, Jovanovic-Talisman T, Rida P, LaBarge M, Seewaldt V. Kinesin Family Member C1 (KIFC1/HSET): A Potential Actionable Biomarker of Early Stage Breast Tumorigenesis and Progression of High-Risk Lesions. Journal of Personalized Medicine. 2021; 11(12):1361. https://doi.org/10.3390/jpm11121361
Chicago/Turabian StyleWright, Nikita, Zhihong Gong, Rick Kittles, Rama Natarajan, Tijana Jovanovic-Talisman, Padmashree Rida, Mark LaBarge, and Victoria Seewaldt. 2021. "Kinesin Family Member C1 (KIFC1/HSET): A Potential Actionable Biomarker of Early Stage Breast Tumorigenesis and Progression of High-Risk Lesions" Journal of Personalized Medicine 11, no. 12: 1361. https://doi.org/10.3390/jpm11121361
APA StyleWright, N., Gong, Z., Kittles, R., Natarajan, R., Jovanovic-Talisman, T., Rida, P., LaBarge, M., & Seewaldt, V. (2021). Kinesin Family Member C1 (KIFC1/HSET): A Potential Actionable Biomarker of Early Stage Breast Tumorigenesis and Progression of High-Risk Lesions. Journal of Personalized Medicine, 11(12), 1361. https://doi.org/10.3390/jpm11121361