
Deep Learning for Integrated Analysis of Insulin Resistance with Multi-Omics Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. iHMP Type 2 Diabetes Mellitus Data Description
2.2. Predictive Models for Insulin Resistance and Insulin Sensitivity (IRIS)
2.3. Backward Elimination for Feature Selection
2.4. Predictive Models for IRIS with Selected Features
2.5. Predictive Models for IRIS with Microbiome Feature Substitution
2.6. Random Sample Permutation
2.7. Deep Neural Network Interpretation Algorithm
2.8. Statistical Analysis
3. Results
3.1. Baseline Characteristics of the iHMP Dataset
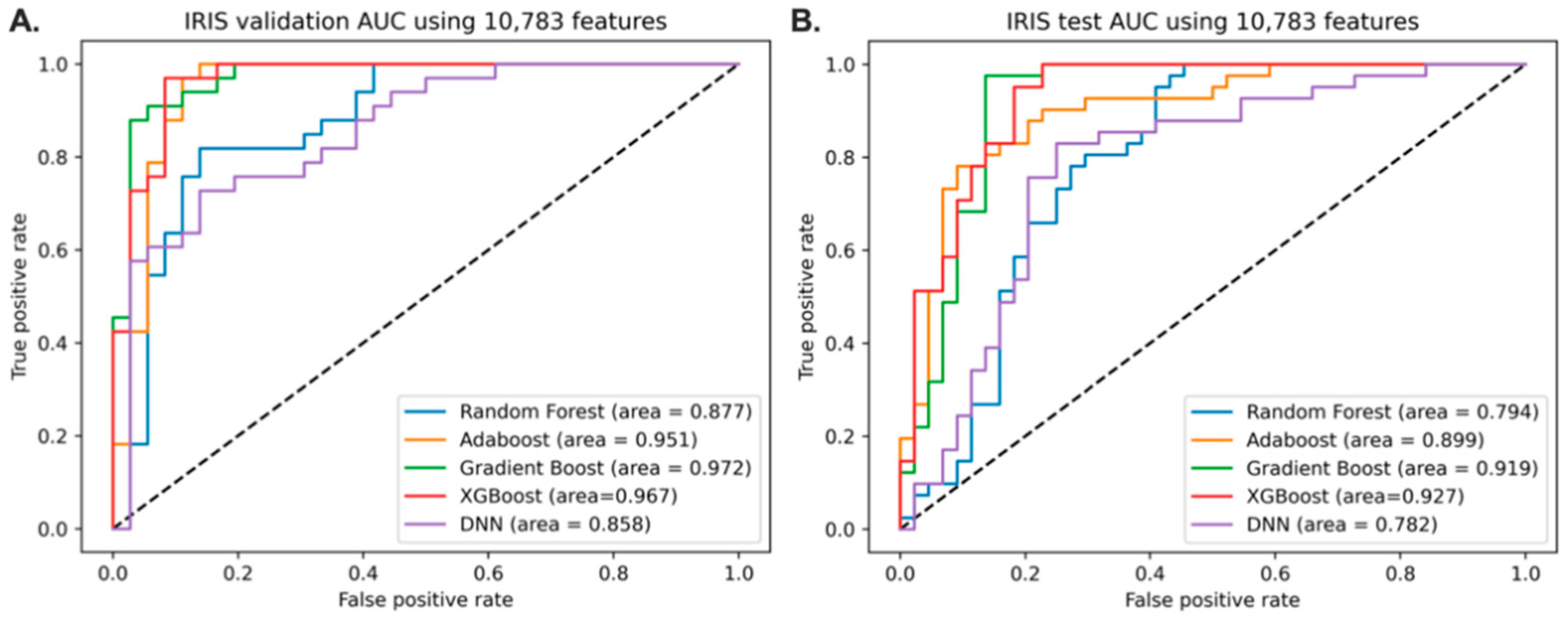
3.2. Predictive Models of IRIS with Full Features
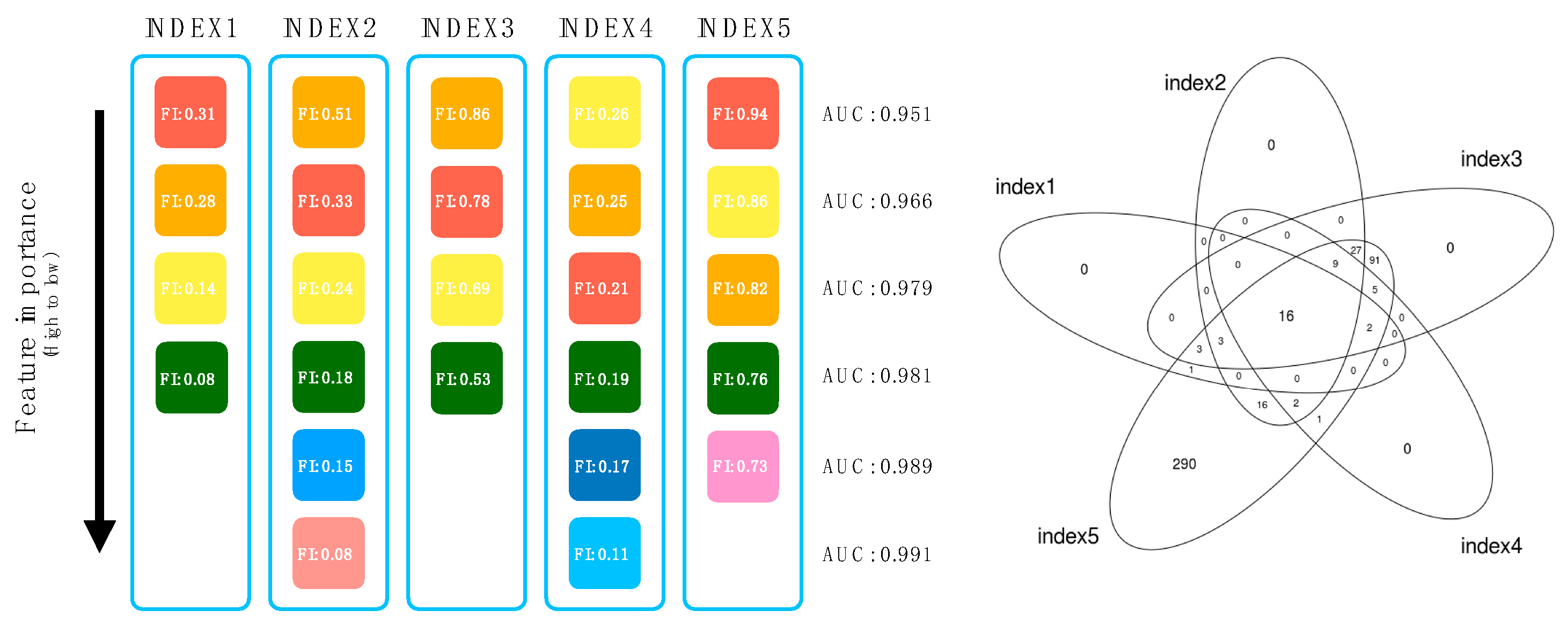
3.3. Backward Elimination for Feature Reduction and Selection
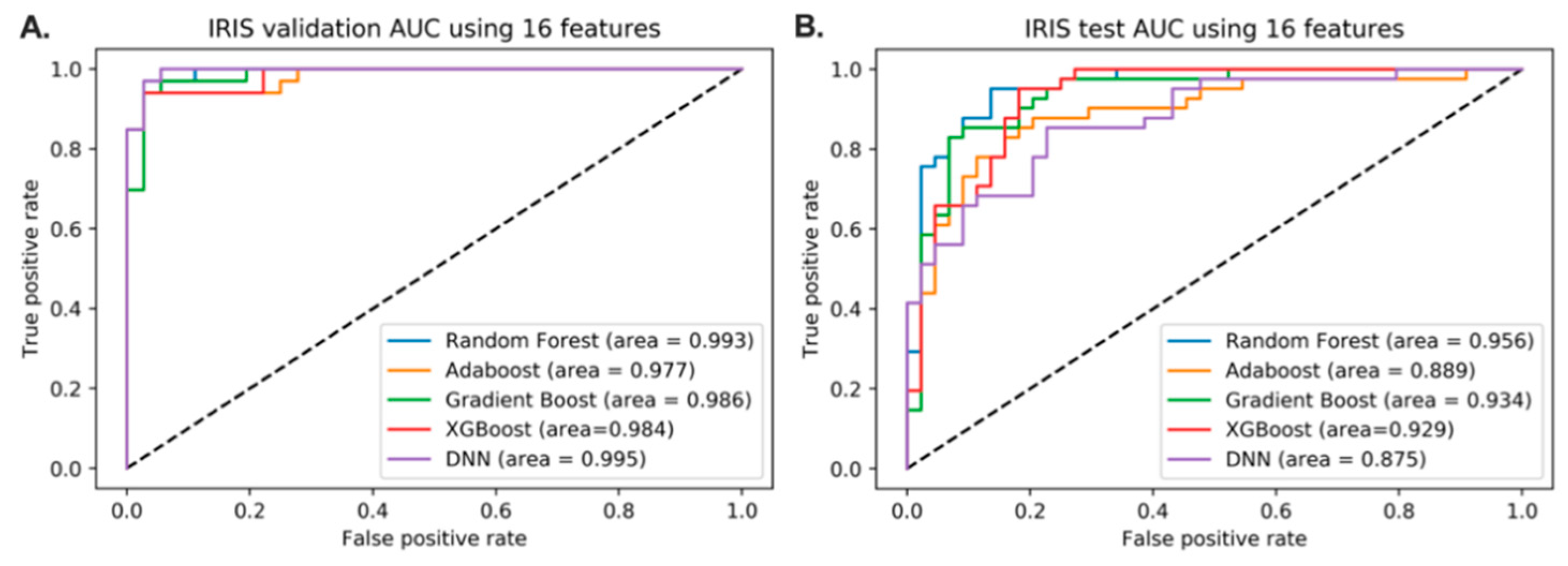
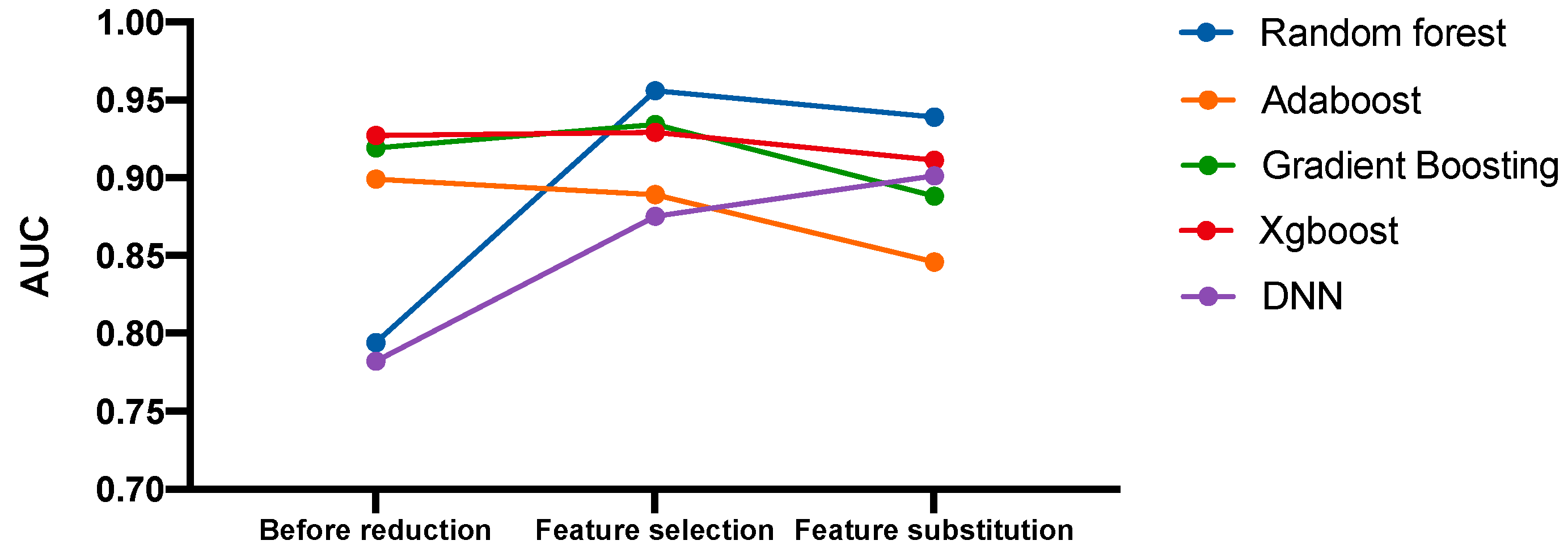
3.4. Predictive Models of IRIS Based on Feature Reduction
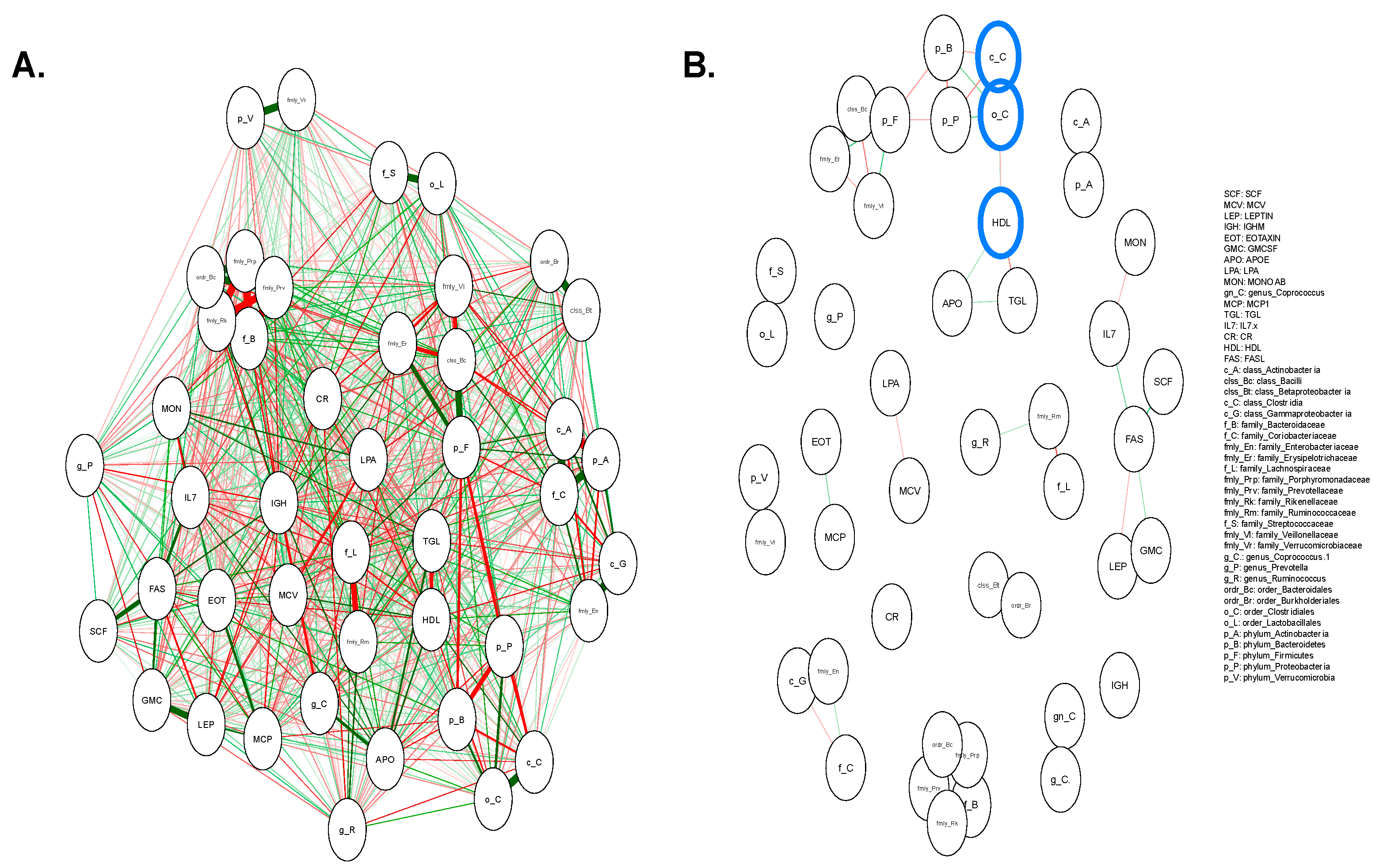
3.5. Pairwise Correlation Network between the Microbiome and Extracted Features
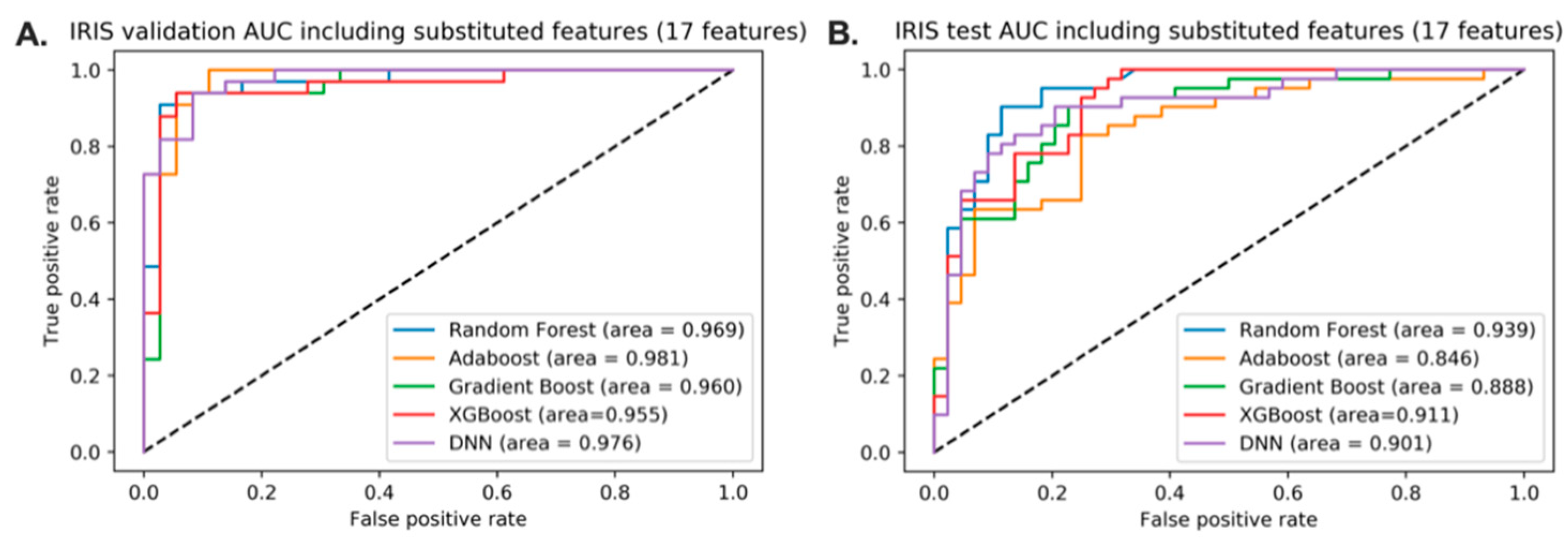
3.6. Predictive Model of IRIS with the Replacement of Corresponding Features with Microbiome Variables
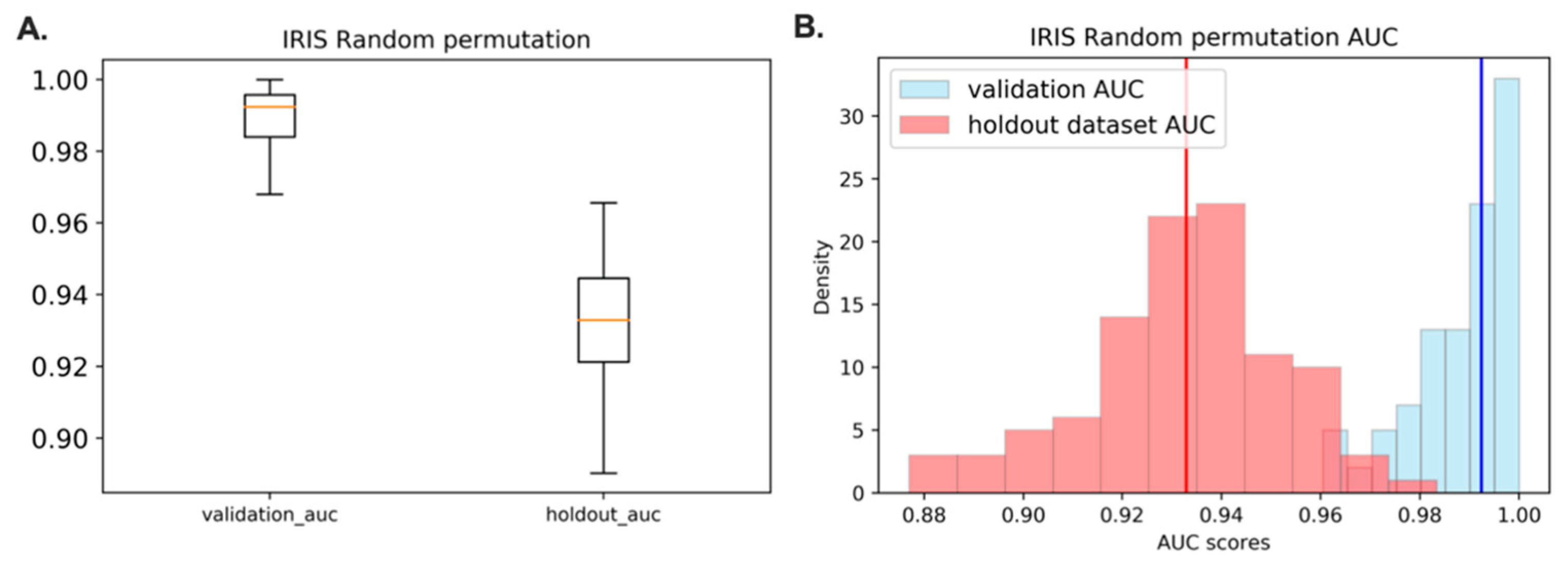
3.7. Acquire the Representative Model with the Highest Frequency in the Random Permutation
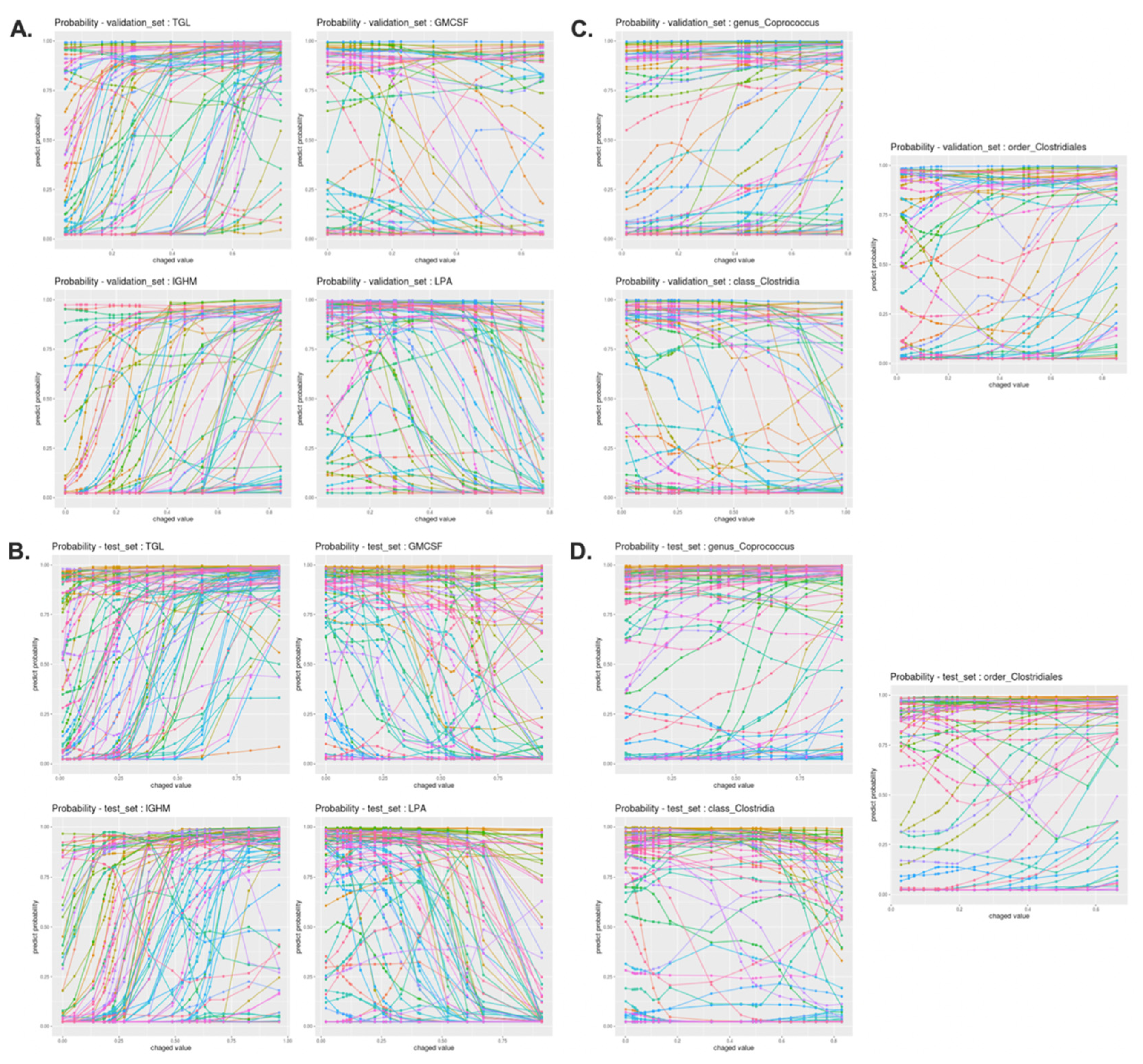
3.8. Interpretation of 17 Features of DNN Classification Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sardaraz, M.; Tahir, M.; Ikram, A.A. Advances in high throughput DNA sequence data compression. J. Bioinform. Comput. Biol. 2016, 14, 1–18. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of Applications of High-Throughput Sequencing in Personalized Medicine: Barriers and Facilitators of Future Progress in Research and Clini-cal Application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; Boucher, C. Sequencing Technologies and Analyses: Where Have We Been and Where Are We Going? iScience 2019, 18, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, Functions and Dynamics in the Expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Proctor, L.M.; Creasy, H.H.; Fettweis, J.M. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Fodor, A.A.; Desantis, T.Z.; Wylie, K.M.; Badger, J.H.; Ye, Y.; Hepburn, T.; Hu, P.; Sodergren, E.; Liolios, K.; Huot-Creasy, H.; et al. The “Most Wanted” Taxa from the Human Microbiome for Whole Genome Sequencing. PLoS ONE 2012, 7, e41294. [Google Scholar] [CrossRef]
- Nelson, K.E.; Weinstock, G.M.; Highlander, S.K.; Worley, K.C.; Creasy, H.H.; Wortman, J.R.; Rusch, D.B.; Mitreva, M.; Sodergren, E.; Chinwalla, A.T.; et al. A Catalog of Reference Ge-nomes from the Human Microbiome. Science 2010, 328, 994–999. [Google Scholar]
- Wylie, K.M.; Truty, R.M.; Sharpton, T.J.; Mihindukulasuriya, K.A.; Zhou, Y.; Gao, H.; Sodergren, E.; Weinstock, G.M.; Pollard, K.S. Novel Bacterial Taxa in the Human Microbiome. PLoS ONE 2012, 7, e35294. [Google Scholar] [CrossRef]
- Li, K.; Bihan, M.; Yooseph, S.; Methé, B.A. Analyses of the Microbial Diversity across the Human Microbiome. PLoS ONE 2012, 7, e32118. [Google Scholar] [CrossRef]
- Zhou, W.; Sailani, M.R.; Contrepois, K.; Zhou, Y.; Ahadi, S.; Leopold, S.R.; Zhang, M.J.; Rao, V.; Avina, M.; Mishra, T.; et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nat. Cell Biol. 2019, 569, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, B.; Tian, T.S.; Soares, J.C. A Review of Feature Reduction Techniques in Neuroimaging. Neuroinformatics 2013, 12, 229–244. [Google Scholar] [CrossRef]
- Kondo, M.; Bezemer, C.-P.; Kamei, Y.; Hassan, A.E.; Mizuno, O. The Impact of Feature Reduction Techniques on Defect Pre-diction Models. Empir. Softw. Eng. 2019, 24, 1925–1963. [Google Scholar] [CrossRef]
- Li, J.; Lu, Q.; Wen, Y. Multi-kernel linear mixed model with adaptive lasso for prediction analysis on high-dimensional multi-omics data. Bioinformatics 2020, 36, 1785–1794. [Google Scholar] [CrossRef]
- Coretto, P.; Serra, A.; Tagliaferri, R. Robust Clustering of Noisy High-Dimensional Gene Expression Data for Patients Sub-typing. Bioinformatics 2018, 34, 4064–4072. [Google Scholar]
- Ho, N.T.; Li, F.; Wang, S.; Kuhn, L. metamicrobiomeR: An R package for analysis of microbiome relative abundance data using zero-inflated beta GAMLSS and meta-analysis across studies using random effects models. BMC Bioinform. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ahn, T.; Goo, T.; Lee, C.-H.; Kim, S.; Han, K.; Park, S.; Park, T. Deep Learning-based Identification of Cancer or Normal Tissue using Gene Expression Data. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 1748–1752. [Google Scholar]
- Li, J.-W.; Li, L.-L.; Chang, L.-L.; Wang, Z.-Y.; Xu, Y. Stem cell factor protects against neuronal apoptosis by activating AKT/ERK in diabetic mice. Braz. J. Med Biol. Res. 2009, 42, 1044–1049. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, K.; Paramel, G.V.; Kienesberger, P.C. Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients 2018, 10, 399. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sandoval, D.; Reed, J.A.; Matter, E.K.; Tolod, E.G.; Woods, S.C.; Seeley, R.J. The Role of GM-CSF in Adipose Tis-sue Inflammation. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1038–E1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, S.; Taront, S.; Magnan, C.; Fauconnier, L.; Delacre, M.; Macia, L.; Delanoye, A.; Verwaerde, C.; Spriet, C.; Saule, P.; et al. Interleukin-7 Regulates Adipose Tissue Mass and Insulin Sensitivity in High-Fat Diet-Fed Mice through Lymphocyte-Dependent and Independent Mechanisms. PLoS ONE 2012, 7, e40351. [Google Scholar] [CrossRef]
- Moon, J.S.; Lee, J.E.; Yoon, J.S. Variation in Serum Creatinine Level Is Correlated to Risk of Type 2 Diabetes. Endocrinol. Metab. 2013, 28, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Katagiri, H.; Ishigaki, Y.; Yamada, T.; Ogihara, T.; Imai, J.; Uno, K.; Hasegawa, Y.; Kanzaki, M.; Yamamoto, T.T.; et al. Involvement of Apolipoprotein E in Excess Fat Accumulation and Insulin Resistance. Diabetes 2006, 56, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Lyngdorf, L.G.; Gregersen, S.; Daugherty, A.; Falk, E. Paradoxical reduction of atherosclerosis in apoE-deficient mice with obesity-related type 2 diabetes. Cardiovasc. Res. 2003, 59, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Schreyer, S.A.; Vick, C.; Lystig, T.C.; Mystkowski, P.; Leboeuf, R.C. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am. J. Physiol. Metab. 2002, 282, E207–E214. [Google Scholar] [CrossRef]
- Lee, E.Y.; Yang, H.K.; Lee, J.; Kang, B.; Yang, Y.; Lee, S.-H.; Ko, S.-H.; Ahn, Y.-B.; Cha, B.Y.; Yoon, K.-H.; et al. Triglyceride glucose index, a marker of insulin resistance, is associated with coronary artery stenosis in asymptomatic subjects with type 2 diabetes. Lipids Health Dis. 2016, 15, 155. [Google Scholar] [CrossRef] [Green Version]
- Kawarabayashi, R.; Motoyama, K.; Nakamura, M.; Yamazaki, Y.; Morioka, T.; Mori, K.; Fukumoto, S.; Imanishi, Y.; Shioi, A.; Shoji, T.; et al. The Association between Monocyte Surface CD163 and Insulin Resistance in Patients with Type 2 Diabetes. J. Diabetes Res. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Harmon, D.B.; Srikakulapu, P.; Kaplan, J.L.; Oldham, S.N.; McSkimming, C.; Garmey, J.C.; Perry, H.M.; Kirby, J.L.; Prohaska, T.A.; Gonen, A.; et al. Protective role for B1b B cells and IgM in obesity-associated inflammation, glucose intolerance, and insulin resistance. Arter. Thromb. Vasc. Biol. 2016, 36, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Kumar, H.; Mishra, M.; Bajpai, S.; Pokhria, D.; Arya, A.K.; Singh, R.K.; Tripathi, K. Correlation of insulin resistance, beta cell function and insulin sensitivity with serum sFas and sFasL in newly diagnosed type 2 diabetes. Acta Diabetol. 2011, 50, 511–518. [Google Scholar] [CrossRef]
- Wang, J.; Obici, S.; Morgan, K.; Barzilai, N.; Feng, Z.; Rossetti, L. Overfeeding Rapidly Induces Leptin and Insulin Resistance. Diabetes 2001, 50, 2786–2791. [Google Scholar] [CrossRef] [Green Version]
- Osegbe, I.; Okpara, H.; Azinge, E. Relationship between serum leptin and insulin resistance among obese Nigerian women. Ann. Afr. Med. 2016, 15, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, A.R.; Wu, H.; Xydakis, A.M.; Jones, P.H.; Smith, E.O.; Sweeney, J.F.; Corry, D.B.; Ballantyne, C.M. Eotaxin and Obesity. J. Clin. Endocrinol. Metab. 2006, 91, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Ziaee, A.; Ghorbani, A.; Kalbasi, S.; Hejrati, A.; Moradi, S. Association of hematological indices with prediabetes: A cross-sectional study. Electron. Phys. 2017, 9, 5206–5211. [Google Scholar] [CrossRef] [Green Version]
- Naderpoor, N.; Mousa, A.; Gomez-Arango, L.F.; Barrett, H.L.; Nitert, M.D.; De Courten, B. Faecal Microbiota Are Related to Insulin Sensitivity and Secretion in Overweight or Obese Adults. J. Clin. Med. 2019, 8, 452. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zheng, J.; Shi, W.; Du, N.; Xu, X.; Zhang, Y.; Ji, P.; Zhang, F.; Jia, Z.; Wang, Y.; et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut 2018, 67, 1614–1625. [Google Scholar] [CrossRef]
- Kuang, Y.-S.; Lu, J.-H.; Li, S.-H.; Li, J.-H.; Yuan, M.-Y.; He, J.-R.; Chen, N.-N.; Xiao, W.-Q.; Shen, S.-Y.; Qiu, L.; et al. Connections between the human gut microbiome and gestational diabetes mellitus. GigaScience 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Allin, K.H.; The IMI-DIRECT Consortium; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Chacón, M.R.; Fernández-Real, J.M.; Richart, C.; Megía, A.; Gómez, J.M.; Miranda, M.; Caubet, E.; Pastor, R.; Masdevall, C.; Vilarrasa, N.; et al. Monocyte Chemoattractant Protein-1 in Obesity and Type 2 Diabetes. Insulin Sensitivity Study*. Obesity 2007, 15, 664–672. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Lin, Y.; Ye, S.; He, Y.; Li, S.; Chen, Y.; Zhai, Z. Short-term insulin intensive therapy decreases MCP-1 and NF-κB expression of peripheral blood monocyte and the serum MCP-1 concentration in newlydiagnosed type 2 diabetics. Arch. Endocrinol. Metab. 2018, 62, 212–220. [Google Scholar] [CrossRef]
- Westerbacka, J.; Corner, A.; Kolak, M.; Makkonen, J.; Turpeinen, U.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Insulin regulation of MCP-1 in human adipose tissue of obese and lean women. Am. J. Physiol. Metab. 2008, 294, E841–E845. [Google Scholar] [CrossRef] [PubMed]
- Evers-van Gogh, I.J.; Oteng, A.-B.; Alex, S.; Hamers, N.; Catoire, M.; Stienstra, R.; Kalkhoven, E.; Kersten, S. Muscle-specific inflammation induced by MCP-1 overexpression does not affect whole-body insulin sensitivity in mice. Diabetologia 2016, 59, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IS (n = 25) | IR (n = 32) | P | ||||
|---|---|---|---|---|---|---|
| Age | 56.144 ± 8.098 | 57.223 ± 7.062 | 0.6 | |||
| BMI | 27.474 ± 3.614 | 29.953 ± 3.558 | 0.013 | |||
| Gender | Male (n = 11) | Male (n = 16) | ||||
| Training set | p | Holdout dataset | p | |||
| IS (n = 179) | IR (n = 164) | IS (n = 44) | IR (n = 41) | |||
| SSPG | 101.083 ± 29.354 | 199.402 ± 35.19 | <0.001 | 105.645 ± 27.412 | 203.422 ± 31.473 | <0.001 |
| GLU | 101.994 ± 17.672 | 92.543 ± 11.954 | <0.001 | 93.386 ± 11.429 | 89.537 ± 10.46 | 0.109 |
| HbA1c | 5.713 ± 0.423 | 5.558 ± 0.359 | <0.001 | 5.666 ± 0.557 | 5.498 ± 0.313 | 0.088 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, E.; Kim, S.; Ahn, T. Deep Learning for Integrated Analysis of Insulin Resistance with Multi-Omics Data. J. Pers. Med. 2021, 11, 128. https://doi.org/10.3390/jpm11020128
Huang E, Kim S, Ahn T. Deep Learning for Integrated Analysis of Insulin Resistance with Multi-Omics Data. Journal of Personalized Medicine. 2021; 11(2):128. https://doi.org/10.3390/jpm11020128
Chicago/Turabian StyleHuang, Eunchong, Sarah Kim, and TaeJin Ahn. 2021. "Deep Learning for Integrated Analysis of Insulin Resistance with Multi-Omics Data" Journal of Personalized Medicine 11, no. 2: 128. https://doi.org/10.3390/jpm11020128
APA StyleHuang, E., Kim, S., & Ahn, T. (2021). Deep Learning for Integrated Analysis of Insulin Resistance with Multi-Omics Data. Journal of Personalized Medicine, 11(2), 128. https://doi.org/10.3390/jpm11020128