
Treatment-Resistant Depression Revisited: A Glimmer of Hope




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endocrinopathies
2.1. HPA Axis
2.1.1. Measurement
2.1.2. Treatment
2.2. Diabetes and Depression
2.3. Gonadal Hormones
Possible Treatments
3. Vitamins
Vitamin D
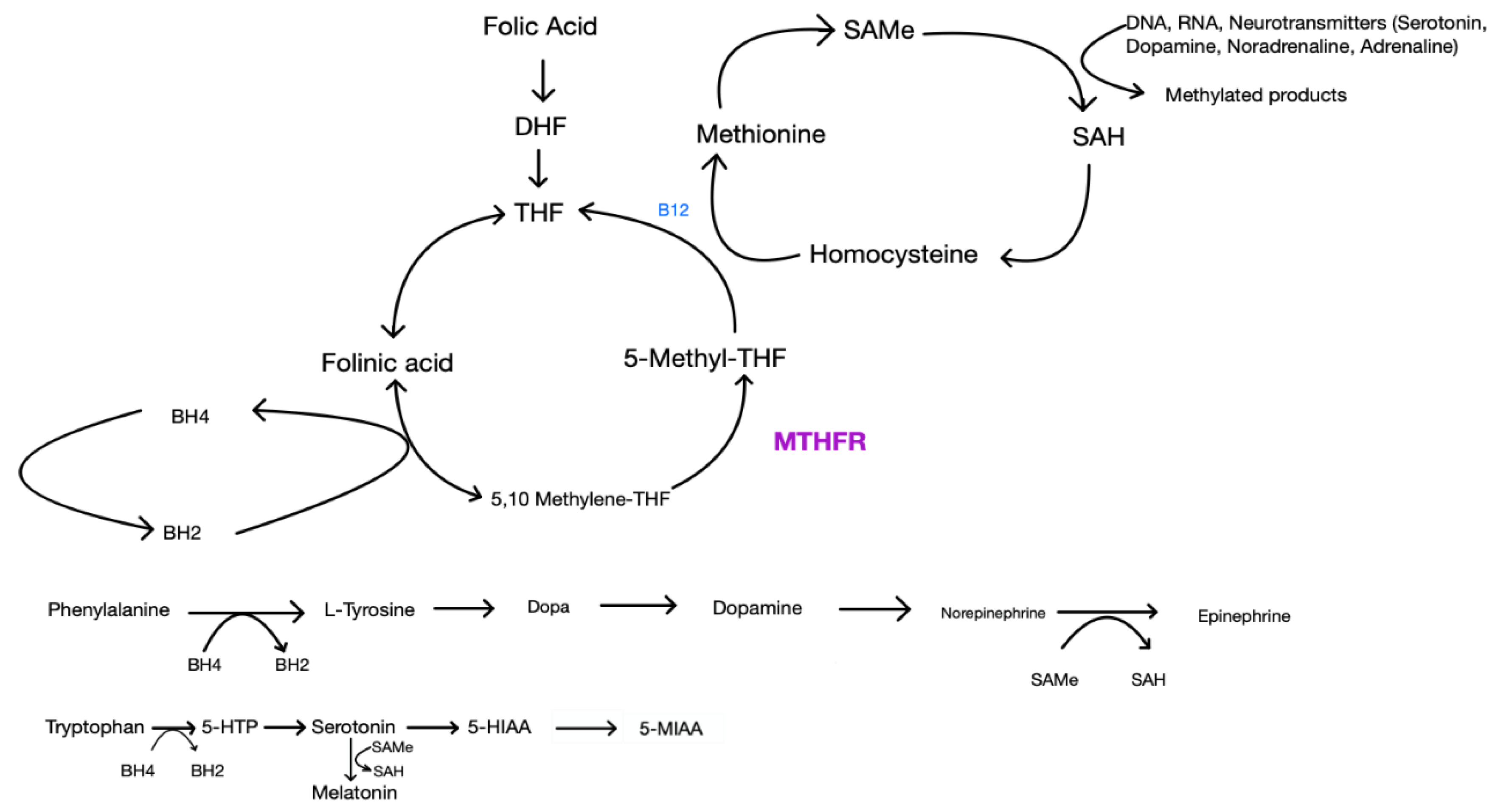
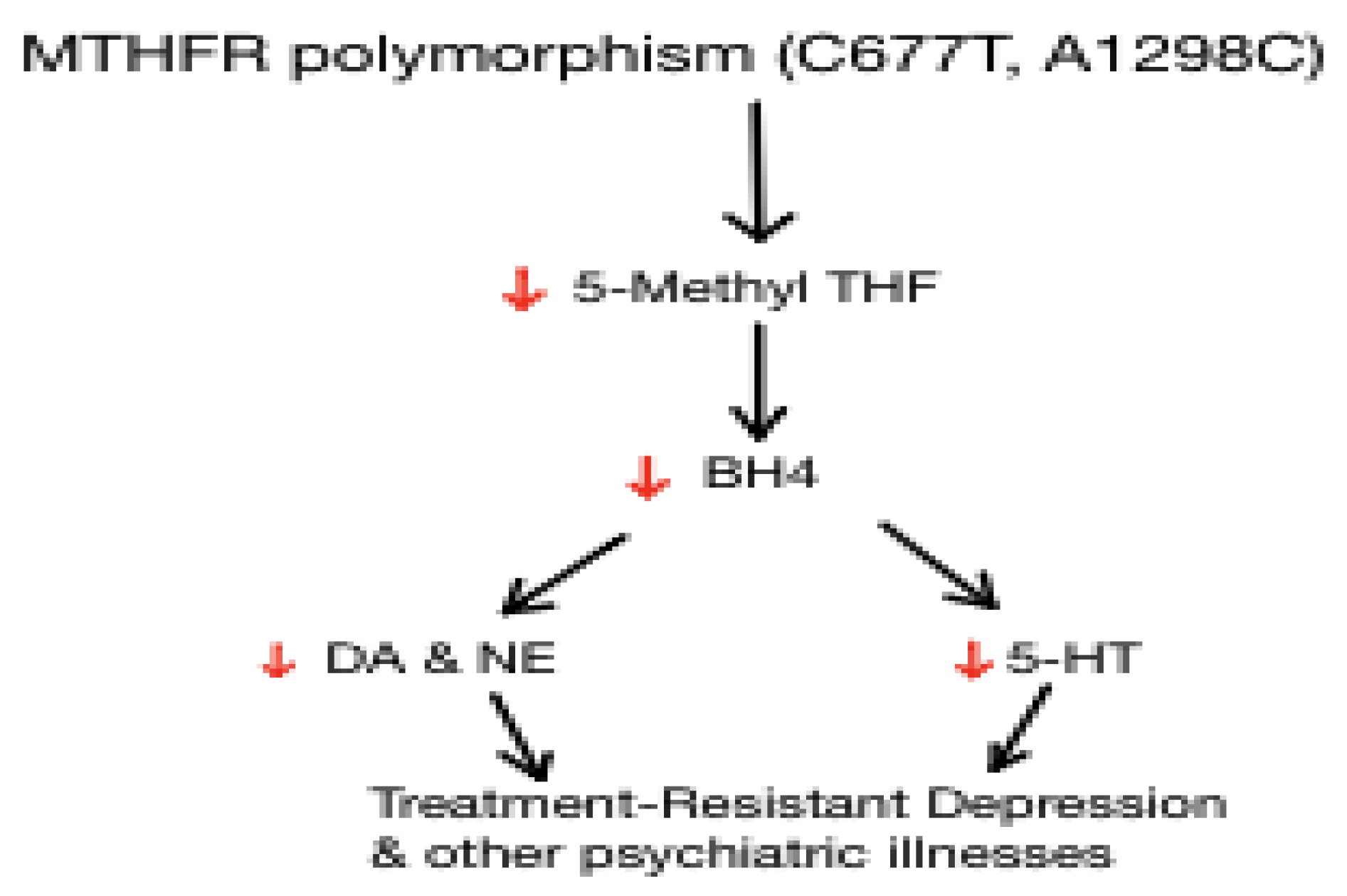
4. Folic acid
Folic Acid, L-methylfolate, MTHFR
5. Pharmacogenomics
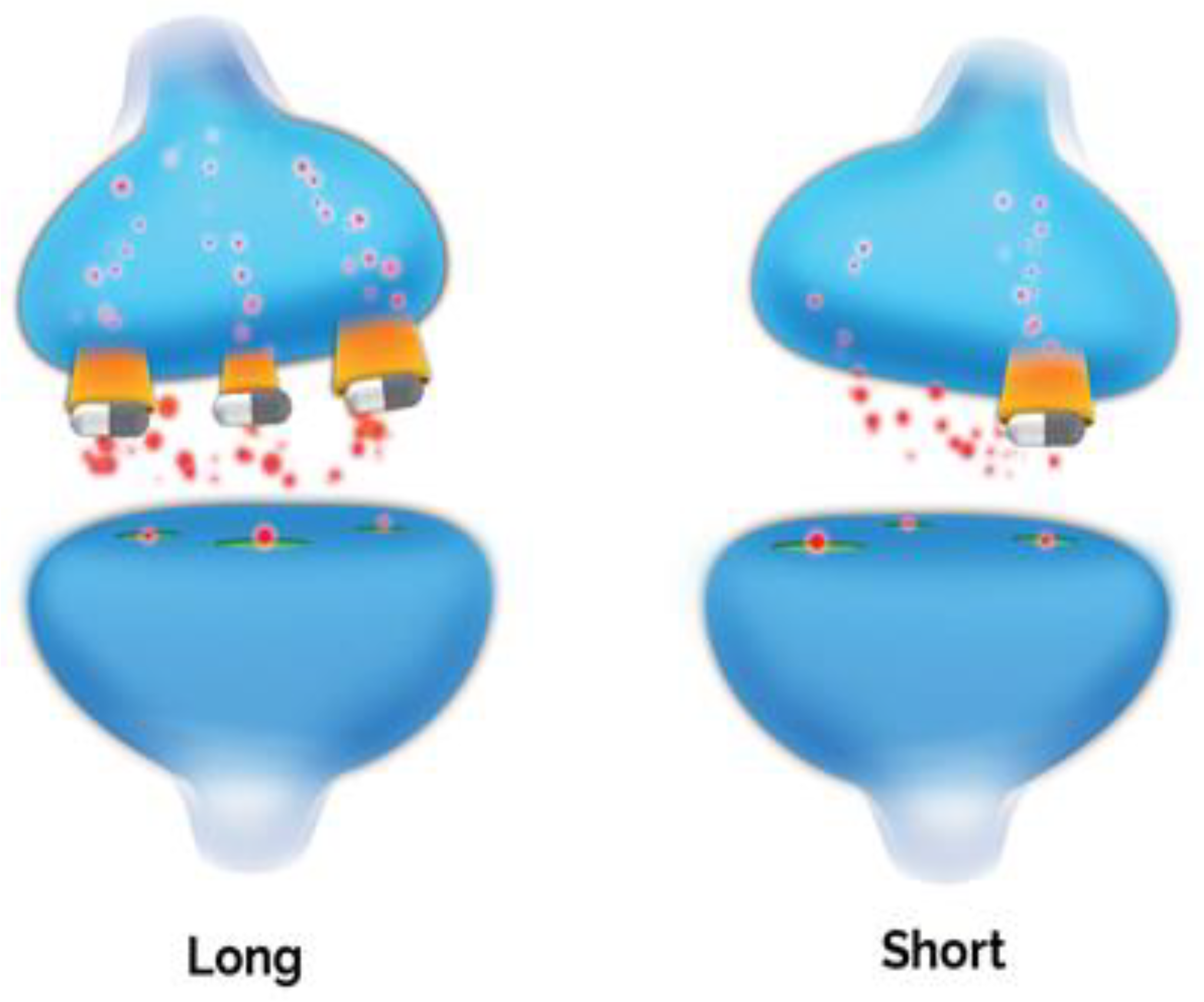
6. The Possible Role of 5-HTTLPR in Neuroprogression in Depression
Serotonin-Transporter-Linked Polymorphic Region (5-HTTLPR)
7. Immune System
7.1. Inflammation and Depression
7.2. C-Reactive Protein (CRP)
7.3. Cytokines
7.4. Anti-Inflammatory Treatments in Affective Disorders
8. Ancillary Treatments
Non-Biologic Factors and Combined Treatment
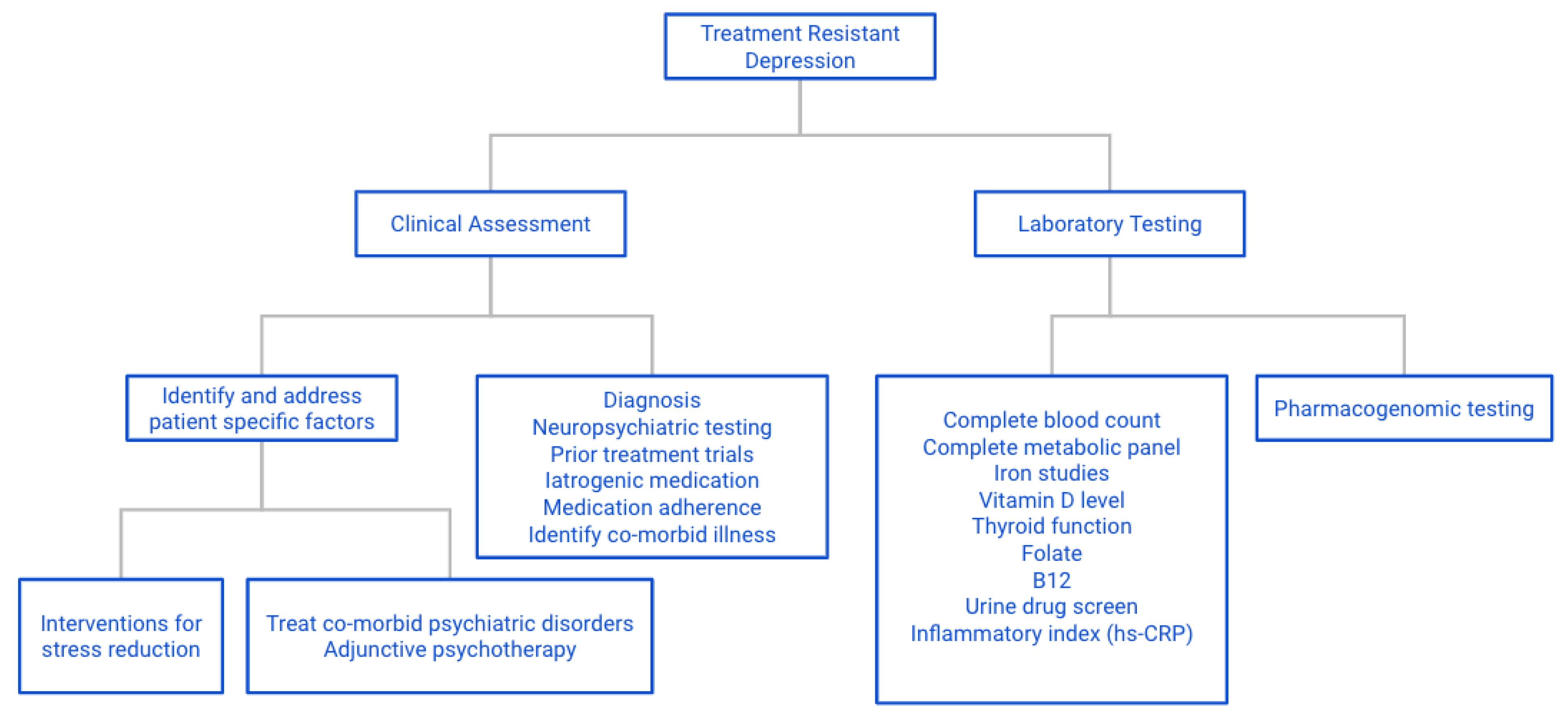
9. Conclusions and Recommendations for Practicing Personalized Medicine
9.1. General
9.2. Specific
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D.; et al. Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef]
- Keitner, G.I.; Mansfield, A.K. Management of Treatment-Resistant Depression. Psychiatry Clin. N. Am. 2012, 35, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B. Augmentation strategies in patients with refractory depression. Depression Anxiety 1996, 4, 169–181. [Google Scholar] [CrossRef]
- Nemeroff, C.B. Use of atypical antipsychotics in refractory depression and anxiety. J. Clin. Psychiatry 2005, 66 (Suppl. S8), 13–21. [Google Scholar]
- Rush, A.; Thase, M.E.; Dubé, S. Research issues in the study of difficult-to-treat depression. Biol. Psychiatry 2003, 53, 743–753. [Google Scholar] [CrossRef]
- Rush, A.J.; Aaronson, S.T.; Demyttenaere, K. Difficult-to-treat depression: A clinical and research roadmap for when remission is elusive. Aust. N. Z. J. Psychiatry 2018, 53, 109–118. [Google Scholar] [CrossRef] [PubMed]
- McAllister-Williams, R.; Arango, C.; Blier, P.; Demyttenaere, K.; Falkai, P.; Gorwood, P.; Hopwood, M.; Javed, A.; Kasper, S.; Malhi, G.; et al. The identification, assessment and management of difficult-to-treat depression: An international consensus statement. J. Affect. Disord. 2020, 267, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Souery, D.; Pitchot, W.; Kasper, S.; Montgomery, S. Definitions and Predictors of Treatment-Resistant Depression. In Treatment—Resistant Depression; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–20. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Hollander, E.; Nutt, D.; Blier, P. Clinical evidence and potential neurobiological underpinnings of unresolved symptoms of depression. J. Clin. Psychiatry 2008, 69, 246–258. [Google Scholar] [CrossRef]
- Zarate, C.; Duman, R.S.; Liu, G.; Sartori, S.; Quiroz, J.; Murck, H. New paradigms for treatment-resistant depression. Ann. N. Y. Acad. Sci. 2013, 1292, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, C.; Porcelli, S.; Serretti, A. Genetics of Treatment-resistant Depression. In Treatment-Resistant Depression; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 43–90. [Google Scholar] [CrossRef]
- Wu, Y.; Mitra, R. Prefrontal-hippocampus plasticity reinstated by an enriched environment during stress. Neurosci. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Monroe, S.M.; Harkness, K.L. Life Stress, the “Kindling” Hypothesis, and the Recurrence of Depression: Considerations from a Life Stress Perspective. Psychol. Rev. 2005, 112, 417–445. [Google Scholar] [CrossRef] [Green Version]
- Lucassen, P.J.; Pruessner, J.; Sousa, N.; Almeida, O.F.X.; Van Dam, A.M.; Rajkowska, G.; Swaab, D.F.; Czéh, B. Neuropathology of stress. Acta Neuropathol. 2013, 127, 109–135. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhao, Y.; Wang, Y.; Liu, L.; Zhang, X.; Li, B.; Cui, R. The Effects of Psychological Stress on Depression. Curr. Neuropharmacol. 2015, 13, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetler, C.; Miller, G.E. Depression and Hypothalamic-Pituitary-Adrenal Activation: A Quantitative Summary of Four Decades of Research. Psychosom. Med. 2011, 73, 114–126. [Google Scholar] [CrossRef]
- Knorr, U.; Vinberg, M.; Kessing, L.V.; Wetterslev, J. Salivary cortisol in depressed patients versus control persons: A systematic review and meta-analysis. Psychoneuroendocrinology 2010, 35, 1275–1286. [Google Scholar] [CrossRef]
- Strawbridge, R.; Young, A.H.; Cleare, A.J. Biomarkers for depression: Recent insights, current challenges and future prospects. Neuropsychiatr. Dis. Treat. 2017, 13, 1245–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, N.; Steffen, J.; Penz, M.; Kirschbaum, C.; Walther, A. Examination of peripheral basal and reactive cortisol levels in major depressive disorder and the burnout syndrome: A systematic review. Neurosci. Biobehav. Rev. 2020, 114, 232–270. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Carter, B.; Marwood, L.; Bandelow, B.; Tsapekos, D.; Nikolova, V.L.; Taylor, R.; Mantingh, T.; De Angel, V.; Patrick, F.; et al. Augmentation therapies for treatment-resistant depression: Systematic review and meta-analysis. Br. J. Psychiatry 2018, 214, 42–51. [Google Scholar] [CrossRef]
- Goodyer, I.M.; Herbert, J.; Tamplin, A.; Altham, P.M.E. Recent life events, cortisol, dehydroepiandrosterone and the onset of major depression in high-risk adolescents. Br. J. Psychiatry 2000, 177, 499–504. [Google Scholar] [CrossRef]
- Harris, T.O.; Borsanyi, S.; Messari, S.; Stanford, K.; Cleary, S.E.; Shiers, H.M.; Brown, G.W.; Herbert, J. Morning cortisol as a risk factor for subsequent major depressive disorder in adult women. Br. J. Psychiatry 2000, 177, 505–510. [Google Scholar] [CrossRef]
- Otte, C.; Wingenfeld, K.; Kuehl, L.K.; Kaczmarczyk, M.; Richter, S.; Quante, A.; Regen, F.; Bajbouj, M.; Zimmermann-Viehoff, F.; Wiedemann, K.; et al. Mineralocorticoid Receptor Stimulation Improves Cognitive Function and Decreases Cortisol Secretion in Depressed Patients and Healthy Individuals. Neuropsychopharmacology 2014, 40, 386–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardet, L.; Petersen, I.; Nazareth, I. Suicidal Behavior and Severe Neuropsychiatric Disorders Following Glucocorticoid Therapy in Primary Care. Am. J. Psychiatry 2012, 169, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Menke, A. Is the HPA Axis as Target for Depression Outdated, or Is There a New Hope? Front. Psychiatry 2019, 10, 101. [Google Scholar] [CrossRef]
- Hodes, G.E.; Kana, V.; Menard, C.; Merad, M.; Russo, S.J. Neuroimmune mechanisms of depression. Nat. Neurosci. 2015, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Macare, C.; Cleare, A.J. Hypothalamic-pituitary-adrenal (HPA) axis functioning as predictor of antidepressant response–Meta-analysis. Neurosci. Biobehav. Rev. 2017, 83, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Clow, A.; Hucklebridge, F.; Stalder, T.; Evans, P.; Thorn, L. The cortisol awakening response: More than a measure of HPA axis function. Neurosci. Biobehav. Rev. 2010, 35, 97–103. [Google Scholar] [CrossRef]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; Van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K.; Turner, M.L.C. Addison’s disease. Clin. Dermatol. 2006, 24, 276–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariante, C.M. Why are depressed patients inflamed? A reflection on 20 years of research on depression, glucocorticoid resistance and inflammation. Eur. Neuropsychopharmacol. 2017, 27, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Lupien, S.J.; Juster, R.-P.; Raymond, C.; Marin, M.-F. The effects of chronic stress on the human brain: From neurotoxicity, to vulnerability, to opportunity. Front. Neuroendocr. 2018, 49, 91–105. [Google Scholar] [CrossRef]
- Ising, M.; Künzel, H.E.; Binder, E.B.; Nickel, T.; Modell, S.; Holsboer, F. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Jani, B.D.; McLean, G.; Nicholl, B.I.; Barry, S.J.E.; Sattar, N.; Mair, F.S.; Cavanagh, J. Risk assessment and predicting outcomes in patients with depressive symptoms: A review of potential role of peripheral blood based biomarkers. Front. Hum. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Papakostas, G.I.; Öngür, D.; Iosifescu, D.V.; Mischoulon, D.; Fava, M. Cholesterol in mood and anxiety disorders: Review of the literature and new hypotheses. Eur. Neuropsychopharmacol. 2004, 14, 135–142. [Google Scholar] [CrossRef]
- Stalder, T.; Kirschbaum, C.; Kudielka, B.M.; Adam, E.K.; Pruessner, J.C.; Wüst, S.; Dockray, S.; Smyth, N.; Evans, P.; Hellhammer, D.H.; et al. Assessment of the cortisol awakening response: Expert consensus guidelines. Psychoneuroendocrinology 2016, 63, 414–432. [Google Scholar] [CrossRef] [PubMed]
- Arana, G.W.; Baldessarini, R.J.; Ornsteen, M. The Dexamethasone Suppression Test for Diagnosis and Prognosis in Psychiatry. Arch. Gen. Psychiatry 1985, 42, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Juruena, M.F.; Pariante, C.M.; Papadopoulos, A.S.; Poon, L.; Lightman, S.; Cleare, A.J. The role of mineralocorticoid receptor function in treatment-resistant depression. J. Psychopharmacol. 2013, 27, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Baes, C.V.W.; Martins, C.M.S.; Tofoli, S.M.D.C.; Juruena, M.F. Early Life Stress in Depressive Patients: HPA Axis Response to GR and MR Agonist. Front. Psychiatry 2014, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, M.; Arfken, C.; Boutros, N. The DEX/CRH test for major depression: A potentially useful diagnostic test. Psychiatry Res. 2013, 208, 131–139. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of Anti-inflammatory Treatment on Depression, Depressive Symptoms, and Adverse Effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Lightman, S.L.; Birnie, M.T.; Conway-Campbell, B.L. Dynamics of ACTH and Cortisol Secretion and Implications for Disease. Endocr. Rev. 2020, 41, 470–490. [Google Scholar] [CrossRef] [Green Version]
- Perlman, K.; Benrimoh, D.; Israel, S.; Rollins, C.; Brown, E.; Tunteng, J.-F.; You, R.; You, E.; Tanguay-Sela, M.; Snook, E.; et al. A systematic meta-review of predictors of antidepressant treatment outcome in major depressive disorder. J. Affect. Disord. 2019, 243, 503–515. [Google Scholar] [CrossRef]
- Pöhlmann, M.L.; Häusl, A.S.; Harbich, D.; Balsevich, G.; Engelhardt, C.; Feng, X.; Breitsamer, M.; Hausch, F.; Winter, G.; Schmidt, M.V. Pharmacological Modulation of the Psychiatric Risk Factor FKBP51 Alters Efficiency of Common Antidepressant Drugs. Front. Behav. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Bendtzen, K.; Nicoletti, F. Pathogenic con-tribution of the Macrophage migration inhibitory factor family to major depressive disorder and emerging tailored therapeutic approaches. J. Affect. Disord. 2020, 263, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Flaster, H.; Bernhagen, J.; Calandra, T.; Bucala, R. The Macrophage Migration Inhibitory Factor-Glucocorticoid Dyad: Regulation of Inflammation and Immunity. Mol. Endocrinol. 2007, 21, 1267–1280. [Google Scholar] [CrossRef]
- Wang, F.-F.; Zhu, L.-A.; Zou, Y.-Q.; Zheng, H.; Wilson, A.; Yang, C.-D.; Shen, N.; Wallace, D.J.; Weisman, M.H.; Chen, S.-L.; et al. New insights into the role and mechanism of macrophage migration inhibitory factor in steroid-resistant patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R103. [Google Scholar] [CrossRef] [Green Version]
- Scantamburlo, G.; Hansenne, M.; Geenen, V.; Legros, J.; Ansseau, M. Additional intranasal oxytocin to escitalopram improves depressive symptoms in resistant depression: An open trial. Eur. Psychiatry 2015, 30, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Nikkheslat, N.; McLaughlin, A.P.; Hastings, C.; Zajkowska, Z.; Nettis, M.A.; Mariani, N.; Enache, D.; Lombardo, G.; Pointon, L.; Cowen, P.J.; et al. Childhood trauma, HPA axis activity and antidepressant response in patients with depression. Brain Behav. Immun. 2020, 87, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Strawbridge, R.; Vives, A.R.H.; Cleare, A.J. Cortisol as a predictor of psychological therapy response in depressive disorders: Systematic review and meta-analysis. Br. J. Psychiatry 2017, 210, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Freedland, K.E.; Clouse, R.E.; Lustman, P.J. The Prevalence of Comorbid Depression in Adults with Diabetes: A meta-analysis. Diabetes Care 2001, 24, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Darwish, L.; Beroncal, E.; Sison, M.V.; Swardfager, W. Depression in people with type 2 diabetes: Current perspectives. Diabetes Metab. Syndr. Obes. Targets Ther. 2018, 11, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeremiah, O.J.; Cousins, G.; Boland, F.; Kirby, B.P.; Ryan, B.K. Evaluation of the effect of insulin sensitivity-enhancing lifestyle- and dietary-related adjuncts on antidepressant treatment response: A systematic review and meta-analysis. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Lin, K.W.; Wroolie, T.E.; Robakis, T.K.; Rasgon, N.L. Adjuvant pioglitazone for unremitted depression: Clinical correlates of treatment response. Psychiatry Res. 2015, 230, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Watson, K.; Nasca, C.; Aasly, L.; McEwen, B.; Rasgon, N. Insulin resistance, an unmasked culprit in depressive disorders: Promises for interventions. Neuropharmacology 2018, 136, 327–334. [Google Scholar] [CrossRef]
- Doyle, T.; Halaris, A.; Rao, M. Shared Neurobiological Pathways Between Type 2 Diabetes and Depressive Symptoms: A Review of Morphological and Neurocognitive Findings. Curr. Diabetes Rep. 2014, 14. [Google Scholar] [CrossRef]
- McMartin, S.E.; Jacka, F.N.; Colman, I. The association between fruit and vegetable consumption and mental health disorders: Evidence from five waves of a national survey of Canadians. Prev. Med. 2013, 56, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Hermanns, N. Subclinical inflammation and depressive symptoms in patients with type 1 and type 2 diabetes. Semin. Immunopathol. 2019, 41, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.D.M.L.E.; Lam, M.P.; Soares, C.N.; Munoz, D.P.; Milev, R.; De Felice, F.G. Insulin Resistance as a Shared Pathogenic Mechanism Between Depression and Type 2 Diabetes. Front. Psychiatry 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Colle, R.; De Larminat, D.; Rotenberg, S.; Hozer, F.; Hardy, P.; Verstuyft, C.; Fève, B.; Corruble, E. Pioglitazone could induce remission in major depression: A meta-analysis. Neuropsychiatr. Dis. Treat. 2016, 13, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Hammen, C. Stress and Depression. Annu. Rev. Clin. Psychol. 2005, 1, 293–319. [Google Scholar] [CrossRef] [Green Version]
- Viau, V. Functional Cross-Talk Between the Hypothalamic-Pituitary-Gonadal and -Adrenal Axes. J. Neuroendocr. 2002, 14, 506–513. [Google Scholar] [CrossRef]
- Acevedo-Rodriguez, A.; Kauffman, A.S.; Cherrington, B.D.; Borges, C.S.; Roepke, T.A.; Laconi, M. Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signalling. J. Neuroendocr. 2018, 30, e12590. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R. Why is depression more prevalent in women? J. Psychiatry Neurosci. 2015, 40, 219–221. [Google Scholar] [CrossRef]
- Mehta, P.H.; Josephs, R.A. Testosterone and cortisol jointly regulate dominance: Evidence for a dual-hormone hypothesis. Horm. Behav. 2010, 58, 898–906. [Google Scholar] [CrossRef]
- Carré, J.M.; Mehta, P.H. Importance of considering testosterone-cortisol interactions in predicting human aggression and dominance. Aggress. Behav. 2011, 37, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Terburg, D.; Morgan, B.; van Honk, J. The testosterone–cortisol ratio: A hormonal marker for proneness to social aggression. Int. J. Law Psychiatry 2009, 32, 216–223. [Google Scholar] [CrossRef]
- Thériault, R.-K.; Perreault, M.L. Hormonal regulation of circuit function: Sex, systems and depression. Biol. Sex Differ. 2019, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Walther, A.; Rice, T.; Kufert, Y.; Ehlert, U. Neuroendocrinology of a Male-Specific Pattern for Depression Linked to Alcohol Use Disorder and Suicidal Behavior. Front. Psychiatry 2017, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Herbert, J. Cortisol and depression: Three questions for psychiatry. Psychol. Med. 2012, 43, 449–469. [Google Scholar] [CrossRef]
- Tozzi, A.; Bellingacci, L.; Pettorossi, V.E. Rapid Estrogenic and Androgenic Neurosteroids Effects in the Induction of Long-Term Synaptic Changes: Implication for Early Memory Formation. Front. Neurosci. 2020, 14, 572511. [Google Scholar] [CrossRef]
- Hillerer, K.M.; Slattery, D.A.; Pletzer, B. Neurobiological mechanisms underlying sex-related differences in stress-related disorders: Effects of neuroactive steroids on the hippocampus. Front. Neuroendocr. 2019, 55, 100796. [Google Scholar] [CrossRef]
- Bristot, G.; Ascoli, B.; Gubert, C.; Panizzutti, B.; Kapczinski, F.; Rosa, A.R. Progesterone and its metabolites as therapeutic targets in psychiatric disorders. Expert Opin. Ther. Targets 2014, 18, 679–690. [Google Scholar] [CrossRef]
- Schweiger, U.; Deuschle, M.; Weber, B.; Korner, A.; Lammers, C.-H.; Schmider, J.; Gotthardt, U.; Heuser, I. Testosterone, Gonadotropin, and Cortisol Secretion in Male Patients With Major Depression. Psychosom. Med. 1999, 61, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Seidman, S.N.; Miyazaki, M.; Roose, S.P. Intramuscular Testosterone Supplementation to Selective Serotonin Reuptake Inhibitor in Treatment-Resistant Depressed Men: Randomized placebo-controlled clinical trial. J. Clin. Psychopharmacol. 2005, 25, 584–588. [Google Scholar] [CrossRef]
- Pope, H.G.; Amiaz, R.; Brennan, B.P.; Orr, G.; Weiser, M.; Kelly, J.F.; Kanayama, G.; Siegel, A.; Hudson, J.I.; Seidman, S.N. Parallel-Group Placebo-Controlled Trial of Testosterone Gel in Men With Major Depressive Disorder Displaying an Incomplete Response to Standard Antidepressant Treatment. J. Clin. Psychopharmacol. 2010, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Marsh, W.K.; Bromberger, J.T.; Crawford, S.L.; Leung, K.; Kravitz, H.M.; Randolph, J.F.; Joffe, H.; Soares, C.N. Lifelong estradiol exposure and risk of depressive symptoms during the transition to menopause and postmenopause. Menopause 2017, 24, 1351–1359. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Yao, Y.; Fu, S.; Zhang, H.; Li, N.; Zhu, Q.; Zhang, F.; Luan, F.; Zhao, Y.; He, Y. The prevalence of depressive symptoms in Chinese longevous persons and its correlation with vitamin D status. BMC Geriatr. 2018, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.; Hirani, V.; Biddulph, J.P. Associations Between Vitamin D Levels and Depressive Symptoms in Later Life: Evidence From the English Longitudinal Study of Ageing (ELSA). J. Gerontol. Ser. A 2017, 73, 1377–1382. [Google Scholar] [CrossRef] [Green Version]
- Milaneschi, Y.; Shardell, M.; Corsi, A.M.; Vazzana, R.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. Serum 25-Hydroxyvitamin D and Depressive Symptoms in Older Women and Men. J. Clin. Endocrinol. Metab. 2010, 95, 3225–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogendijk, W.J.G.; Lips, P.; Dik, M.G.; Deeg, D.J.H.; Beekman, A.T.F.; Penninx, B.W.J.H. Depression Is Associated With Decreased 25-Hydroxyvitamin D and Increased Parathyroid Hormone Levels in Older Adults. Arch. Gen. Psychiatry 2008, 65, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Von Känel, R.; Fardad, N.; Steurer, N.; Horak, N.; Hindermann, E.; Fischer, F.; Gessler, K. Vitamin D Deficiency and Depressive Symptomatology in Psychiatric Patients Hospitalized with a Current Depressive Episode: A Factor Analytic Study. PLoS ONE 2015, 10, e0138550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Högberg, G.; Gustafsson, S.A.; Hällström, T.; Gustafsson, T.; Klawitter, B.; Petersson, M. Depressed adolescents in a case-series were low in vitamin D and depression was ameliorated by vitamin D supplementation. Acta Paediatr. 2012, 101, 779–783. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Zhang, Z.-H. Risk factors for postpartum depression: An evidence-based systematic review of systematic reviews and meta-analyses. Asian J. Psychiatry 2020, 102353. [Google Scholar] [CrossRef] [PubMed]
- Accortt, E.E.; Schetter, C.D.; Peters, R.M.; Cassidy-Bushrow, A.E. Lower prenatal vitamin D status and postpartum depressive symptomatology in African American women: Preliminary evidence for moderation by inflammatory cytokines. Arch. Women’s Ment. Health 2015, 19, 373–383. [Google Scholar] [CrossRef] [Green Version]
- McCann, J.C.; Ames, B.N. Is there convincing biological or behavioral evidence linking vitamin D deficiency to brain dysfunction? FASEB J. 2007, 22, 982–1001. [Google Scholar] [CrossRef]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the Vitamin D receptor and 1α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef]
- Parker, G.B.; Brotchie, H.; Graham, R.K. Vitamin D and depression. J. Affect. Disord. 2017, 208, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gowda, U.; Mutowo, M.P.; Smith, B.J.; Wluka, A.E.; Renzaho, A.M. Vitamin D supplementation to reduce depression in adults: Meta-analysis of randomized controlled trials. Nutrition 2015, 31, 421–429. [Google Scholar] [CrossRef]
- Shaffer, J.A.; Edmondson, D.; Wasson, L.T.; Falzon, L.; Homma, K.; Ezeokoli, N.; Li, P.; Davidson, K.W. Vitamin D Supplementation for Depressive Symptoms: A systematic review and meta-analysis of randomized controlled trials. Psychosom. Med. 2014, 76, 190–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spedding, S. Vitamin D and Depression: A Systematic Review and Meta-Analysis Comparing Studies with and without Biological Flaws. Nutrients 2014, 6, 1501–1518. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Vellekkatt, F. Efficacy of vitamin D supplementation in major depression: A meta-analysis of randomized controlled trials. J. Postgrad. Med. 2018, 65, 74–80. [Google Scholar] [CrossRef]
- Okereke, O.I.; Reynolds, C.F.; Mischoulon, D.; Chang, G.; Vyas, C.M.; Cook, N.R.; Weinberg, A.; Bubes, V.; Copeland, T.; Friedenberg, G.; et al. Effect of Long-term Vitamin D3 Supplementation vs Placebo on Risk of Depression or Clinically Relevant Depressive Symptoms and on Change in Mood Scores: A Randomized Clinical Trial. JAMA 2020, 324, 471. [Google Scholar] [CrossRef]
- Amrein, K.; Scherkl, M.; Hoffmann, M.; Neuwersch-Sommeregger, S.; Köstenberger, M.; Berisha, A.T.; Martucci, G.; Pilz, S.; Malle, O. Vitamin D deficiency 2.0: An update on the current status worldwide. Eur. J. Clin. Nutr. 2020, 74, 1498–1513. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Ionescu, D.F. Towards new mechanisms: An update on therapeutics for treatment-resistant major depressive disorder. Mol. Psychiatry 2015, 20, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Alpert, J.E.; Mischoulon, D.; Nierenberg, A.A.; Fava, M. Nutrition and depression: Focus on folate. Nutrition 2000, 16, 544–546. [Google Scholar] [CrossRef]
- Gilbody, S.; Lewis, S.; Lightfoot, T. Methylenetetrahydrofolate Reductase (MTHFR) Genetic Polymorphisms and Psychiatric Disorders: A HuGE Review. Am. J. Epidemiol. 2006, 165, 1–13. [Google Scholar] [CrossRef]
- Wan, L.; Li, Y.; Zhang, Z.; Sun, Z.; He, Y.; Li, R. Methylenetetrahydrofolate reductase and psychiatric diseases. Transl. Psychiatry 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, genes, polymorphisms and the associated diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef]
- Frankenburg, F.R. The Role of One-Carbon Metabolism in Schizophrenia and Depression. Harv. Rev. Psychiatry 2007, 15, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Peerbooms, O.L.; van Os, J.; Drukker, M.; Kenis, G.; Hoogveld, L.; de Hert, M.; Delespaul, P.; van Winkel, R.; Rutten, B.P. Meta-analysis of MTHFR gene variants in schizophrenia, bipolar disorder and unipolar depressive disorder: Evidence for a common genetic vulnerability? Brain Behav. Immun. 2011, 25, 1530–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, V. Association of C677T polymorphism (rs1801133) in MTHFR gene with depression. Cell. Mol. Biol. 2017, 63, 60–67. [Google Scholar] [CrossRef]
- Duprey, R.P. MTHFR Gene Polymorphism Positive Treatment-Resistant Depression: Prevalence and Treatment Recommendations. Neuropsychiatry 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Sarris, J.; Murphy, J.; Mischoulon, D.; Papakostas, G.I.; Fava, M.; Berk, M.; Ng, C.H. Adjunctive Nutraceuticals for Depression: A Systematic Review and Meta-Analyses. Am. J. Psychiatry 2016, 173, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papakostas, G.I.; Shelton, R.C.; Zajecka, J.M.; Etemad, B.; Rickels, K.; Clain, A.; Baer, L.; Dalton, E.D.; Sacco, G.R.; Schoenfeld, D.; et al. l-Methylfolate as Adjunctive Therapy for SSRI-Resistant Major Depression: Results of Two Randomized, Double-Blind, Parallel-Sequential Trials. Am. J. Psychiatry 2012, 169, 1267–1274. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Shelton, R.C.; Zajecka, J.M.; Bottiglieri, T.; Roffman, J.; Cassiello, C.; Stahl, S.M.; Fava, M. Effect of Adjunctivel-Methylfolate 15 mg Among Inadequate Responders to SSRIs in Depressed Patients Who Were Stratified by Biomarker Levels and Genotype. J. Clin. Psychiatry 2014, 75, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Coppen, A.; Bailey, J. Enhancement of the antidepressant action of fluoxetine by folic acid: A randomised, placebo controlled trial. J. Affect. Disord. 2000, 60, 121–130. [Google Scholar] [CrossRef]
- Resler, G.; Lavie, R.; Campos, J.; Mata, S.; Urbina, M.; García, A.; Apitz, R.; Lima, L. Effect of Folic Acid Combined with Fluoxetine in Patients with Major Depression on Plasma Homocysteine and Vitamin B12, and Serotonin Levels in Lymphocytes. Neuroimmunomodulation 2008, 15, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Shelton, R.C.; Manning, J.S.; Barrentine, L.W.; Tipa, E.V. Assessing Effects of l-Methylfolate in Depression Management. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Owen, R.T. Folate augmentation of antidepressant response. Drugs Today 2013, 49, 791–798. [Google Scholar] [CrossRef]
- Pan, L.A.; Martin, P.; Zimmer, T.; Segreti, A.M.; Kassiff, S.; McKain, B.W.; Baca, C.A.; Rengasamy, M.; Hyland, K.; Walano, N.; et al. Neurometabolic Disorders: Potentially Treatable Abnormalities in Patients With Treatment-Refractory Depression and Suicidal Behavior. Am. J. Psychiatry 2017, 174, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, P.; Toone, B.; Bottiglien, T.; Laundy, M.; Reynolds, E.; Carney, M.; Flynn, T.; Chanarin, I. Enhancement of recovery from psychiatric illness by methylfolate. Lancet 1990, 336, 392–395. [Google Scholar] [CrossRef]
- Roberts, E.; Carter, B.; Young, A.H. Caveat emptor: Folate in unipolar depressive illness, a systematic review and meta-analysis. J. Psychopharmacol. 2018, 32, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Dartois, L.L.; Stutzman, D.L.; Morrow, M. L-methylfolate Augmentation to Antidepressants for Adolescents with Treatment-Resistant Depression: A Case Series. J. Child Adolesc. Psychopharmacol. 2019, 29, 386–391. [Google Scholar] [CrossRef] [Green Version]
- King, W.D.; Ho, V.; Dodds, L.; Perkins, S.L.; Casson, R.I.; Massey, T.E. Relationships among biomarkers of one-carbon metabolism. Mol. Biol. Rep. 2012, 39, 7805–7812. [Google Scholar] [CrossRef]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otaegui-Arrazola, A.; Amiano, P.; Elbusto, A.; Urdaneta, E.; Martínez-Lage, P. Diet, cognition, and Alzheimer’s disease: Food for thought. Eur. J. Nutr. 2013, 53, 1–23. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K.; Carney, M.W.P.; Reynolds, E.H. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Almeida, O.P.; Ford, A.H.; Flicker, L. Systematic review and meta-analysis of randomized placebo-controlled trials of folate and vitamin B12 for depression. Int. Psychogeriatr. 2015, 27, 727–737. [Google Scholar] [CrossRef]
- Ionescu, D.F.; Rosenbaum, J.F.; Alpert, J.E. Pharmacological approaches to the challenge of treatment-resistant depression. Dialogues Clin. Neurosci. 2015, 17, 111–126. [Google Scholar] [PubMed]
- Bhatia, P.; Singh, N. Homocysteine excess: Delineating the possible mechanism of neurotoxicity and depression. Fundam. Clin. Pharmacol. 2015, 29, 522–528. [Google Scholar] [CrossRef]
- Reynolds, E. The Neurology of Folic Acid Deficiency, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 120, pp. 927–943. [Google Scholar] [CrossRef]
- Cantoni, G.L. The nature of the active methyl donor formed enzymatically from l-methionine and adenosinetriphosphate1, 2. J. Am. Chem. Soc. 1952, 74, 2942–2943. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Laundy, M.; Martin, R.; Carney, M.; Nissenbaum, H.; Toone, B.; Johnson, A.; Reynolds, E. S-adenosylmethionine influences monoamine metabolism. Lancet 1984, 324, 224. [Google Scholar] [CrossRef]
- Otero-Losada, M.E.; Rubio, M.C. Acute changes in 5-HT metabolism after S-adenosyl-l-methionine administration. Gen. Pharmacol. Vasc. Syst. 1989, 20, 403–406. [Google Scholar] [CrossRef]
- Curcio, M.; Catto, E.; Stramentinoli, G.; Algeri, S. Effect of S-adenosyl-L-methionine on serotonin metabolism in rat brain. Prog. Neuro-Psychopharmacol. 1978, 2, 65–71. [Google Scholar] [CrossRef]
- Liśkiewicz, P.; Kaczmarczyk, M.; Misiak, B.; Wroński, M.; Bąba-Kubiś, A.; Skonieczna-Żydecka, K.; Marlicz, W.; Bieńkowski, P.; Misera, A.; Pełka-Wysiecka, J.; et al. Analysis of gut microbiota and intestinal integrity markers of inpatients with major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106. [Google Scholar] [CrossRef]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D. S-adenosylmethionine (SAMe) for neuropsychiatric disorders: A clinician-oriented review of research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galizia, I.; Oldani, L.; MacRitchie, K.; Amari, E.; Dougall, D.; Jones, T.N.; Lam, R.W.; Massei, G.J.; Yatham, L.N.; Young, A.H. S-adenosyl methionine (SAMe) for depression in adults. Cochrane Database Syst. Rev. 2016, 10, CD011286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arandjelovic, K.; Eyre, H.A.; Lenze, E.; Singh, A.B.; Berk, M.; Bousman, C. The role of depression pharmacogenetic decision support tools in shared decision making. J. Neural Transm. 2017, 126, 87–94. [Google Scholar] [CrossRef]
- Bousman, C.A.; Arandjelovic, K.; Mancuso, S.G.; Eyre, H.A.; Dunlop, B.W. Pharmacogenetic tests and depressive symptom remission: A meta-analysis of randomized controlled trials. Pharmacogenomics 2019, 20, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblat, J.D.; Lee, Y.; McIntyre, R.S. The effect of pharmacogenomic testing on response and remission rates in the acute treatment of major depressive disorder: A meta-analysis. J. Affect. Disord. 2018, 241, 484–491. [Google Scholar] [CrossRef]
- Brown, L.; Eum, S.; Haga, S.B.; Strawn, J.R.; Zierhut, H. Clinical Utilization of Pharmacogenetics in Psychiatry—Perspectives of Pharmacists, Genetic Counselors, Implementation Science, Clinicians, and Industry. Pharmacopsychiatry 2019, 53, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Hallflavin, D.K.; Winner, J.G.; Allen, J.D.; Jordan, J.; Nesheim, R.S.; Snyder, K.; Drews, M.S.; Eisterhold, L.L.; Biernacka, J.M.; Mrazek, D.A. Using a pharmacogenomic algorithm to guide the treatment of depression. Transl. Psychiatry 2012, 2, e172. [Google Scholar] [CrossRef] [Green Version]
- Hall-Flavin, D.K.; Winner, J.G.; Allen, J.D.; Carhart, J.M.; Proctor, B.; Snyder, K.A.; Drews, M.S.; Eisterhold, L.L.; Geske, J.; Mrazek, D.A. Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet. Genom. 2013, 23, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, J.G.; Carhart, J.M.; Altar, C.A.; Allen, J.D.; DeChairo, B.M. A prospective, randomized, double-blind study assessing the clinical impact of integrated pharmacogenomic testing for major depressive disorder. Discov. Med. 2013, 16, 219–227. [Google Scholar] [PubMed]
- Winner, J.G.; Allen, J.D.; Altar, C.A.; Spahic-Mihajlovic, A. Psychiatric pharmacogenomics predicts health resource utilization of outpatients with anxiety and depression. Transl. Psychiatry 2013, 3, e242. [Google Scholar] [CrossRef]
- Winner, J.G.; Carhart, J.M.; Altar, C.A.; Goldfarb, S.; Allen, J.D.; Lavezzari, G.; Parsons, K.K.; Marshak, A.G.; Garavaglia, S.; DeChairo, B.M. Combinatorial pharmacogenomic guidance for psychiatric medications reduces overall pharmacy costs in a 1 year prospective evaluation. Curr. Med. Res. Opin. 2015, 31, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, M.R.; King, N.; Wang, Y.; Winner, J.G.; Watterson, L.R.; Gunselman, S.; DeChairo, B.M. Analytical validation of a psychiatric pharmacogenomic test. Pers. Med. 2018, 15, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Thase, M.E.; Parikh, S.V.; Rothschild, A.J.; Dunlop, B.W.; Debattista, C.; Conway, C.R.; Forester, B.P.; Mondimore, F.M.; Shelton, R.C.; Macaluso, M.; et al. Impact of Pharmacogenomics on Clinical Outcomes for Patients Taking Medications With Gene-Drug Interactions in a Randomized Controlled Trial. J. Clin. Psychiatry 2019, 80. [Google Scholar] [CrossRef] [Green Version]
- Greden, J.F.; Parikh, S.V.; Rothschild, A.J.; Thase, M.E.; Dunlop, B.W.; DeBattista, C.; Conway, C.R.; Forester, B.P.; Mondimore, F.M.; Shelton, R.C.; et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: A large, patient- and rater-blinded, randomized, controlled study. J. Psychiatr. Res. 2019, 111, 59–67. [Google Scholar] [CrossRef]
- Altar, C.A.; Carhart, J.; Allen, J.D.; Hall-Flavin, D.; Winner, J.; DeChairo, B. Clinical Utility of Combinatorial Pharmacogenomics-Guided Antidepressant Therapy: Evidence from Three Clinical Studies. Mol. Neuropsychiatry 2015, 1, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.; Vranjkovic, O.; Li, J.; Yu, K.; Al Habbab, T.; Johnson, H.; Brown, K.; Jablonski, M.R.; Dechairo, B. The clinical utility of combinatorial pharmacogenomic testing for patients with depression: A meta-analysis. Pharmacogenomics 2020, 21, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Heils, A.; Teufel, A.; Petri, S.; Stöber, G.; Riederer, P.; Bengel, D.; Lesch, K.P. Allelic Variation of Human Serotonin Transporter Gene Expression. J. Neurochem. 2002, 66, 2621–2624. [Google Scholar] [CrossRef]
- Iurescia, S.; Seripa, D.; Rinaldi, M. Looking Beyond the 5-HTTLPR Polymorphism: Genetic and Epigenetic Layers of Regulation Affecting the Serotonin Transporter Gene Expression. Mol. Neurobiol. 2016, 54, 8386–8403. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.-P.; Bengel, D.; Heils, A.; Sabol, S.Z.; Greenberg, B.D.; Petri, S.; Benjamin, J.; Müller, C.R.; Hamer, D.H.; Murphy, D.L. Association of Anxiety-Related Traits with a Polymorphism in the Serotonin Transporter Gene Regulatory Region. Science 1996, 274, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Kunugi, H.; Hattori, M.; Kato, T.; Tatsumi, M.; Sakai, T.; Sasaki, T.; Hirose, T.; Nanko, S. Serotonin transporter gene polymorphisms: Ethnic difference and possible association with bipolar affective disorder. Mol. Psychiatry 1997, 2, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, K.M.; Smits, L.J.M.; Schouten, J.S.A.G.; Stelma, F.F.; Nelemans, P.; Prins, M.H. Influence of SERTPR and STin2 in the serotonin transporter gene on the effect of selective serotonin reuptake inhibitors in depression: A systematic review. Mol. Psychiatry 2004, 9, 433–441. [Google Scholar] [CrossRef]
- Serretti, A.; Calati, R.; Mandelli, L.; De Ronchi, D. Serotonin transporter gene variants and behavior: A comprehensive review. Curr. Drug Targets 2006, 7, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of Life Stress on Depression: Moderation by a Polymorphism in the 5-HTT Gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Culverhouse, R.C.; Saccone, N.L.; Bierut, L.J. The state of knowledge about the relationship between 5-HTTLPR, stress, and depression. J. Affect. Disord. 2018, 228, 205–206. [Google Scholar] [CrossRef]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonda, X.; Hullám, G.; Antal, P.; Eszlari, N.; Petschner, P.; Hökfelt, T.G.; Anderson, I.M.; Deakin, J.F.W.; Juhasz, G.; Bagdy, G. Significance of risk polymorphisms for depression depends on stress exposure. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Gonda, X.; Eszlari, N.; Anderson, I.; Deakin, B.; Juhasz, G.; Bagdy, G. 5-HTTLPR ‘social sensitivity’ short allele may protect against depression after exposure to social network stressors in young people. Eur. Neuropsychopharmacol. 2019, 29, S581–S582. [Google Scholar] [CrossRef]
- Gonda, X.; Eszlari, N.; Kovacs, D.; Anderson, I.M.; Deakin, J.F.W.; Juhász, G.; Bagdy, G. Financial difficulties but not other types of recent negative life events show strong interactions with 5-HTTLPR genotype in the development of depressive symptoms. Transl. Psychiatry 2016, 6, e798. [Google Scholar] [CrossRef] [Green Version]
- Serretti, A.; Kato, M.; De Ronchi, D.; Kinoshita, T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol. Psychiatry 2006, 12, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, S.; Fabbri, C.; Serretti, A. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with antidepressant efficacy. Eur. Neuropsychopharmacol. 2012, 22, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ma, Y.; Zhu, X.; Guo, R.; Wang, J.; He, L. Pharmacogenetic association of bi- and triallelic polymorphisms of SLC6A4 with antidepressant response in major depressive disorder. J. Affect. Disord. 2020, 273, 254–264. [Google Scholar] [CrossRef]
- Gressier, F.; Bouaziz, E.; Verstuyft, C.; Hardy, P.; Becquemont, L.; Corruble, E. 5-HTTLPR modulates antidepressant efficacy in depressed women. Psychiatr. Genet. 2009, 19, 195–200. [Google Scholar] [CrossRef]
- Huezo-Diaz, P.; Uher, R.; Smith, R.P.; Rietschel, M.; Henigsberg, N.; Marušiˇ, A.; Mors, O.; Maier, W.; Hauser, J.W.; Souery, D.; et al. Moderation of antidepressant response by the serotonin transporter gene. Br. J. Psychiatry 2009, 195, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Karlović, D.; Karlović, D. Serotonin transporter gene (5-HTTLPR) polymorphism and efficacy of selective serotonin reuptake inhibitors—Do we have sufficient evidence for clinical practice. Acta Clin. Croat. 2013, 52, 353–362. [Google Scholar] [PubMed]
- Zhu, J.; Klein-Fedyshin, M.; Stevenson, J.M. Serotonin Transporter Gene Polymorphisms and Selective Serotonin Reuptake Inhibitor Tolerability: Review of Pharmacogenetic Evidence. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-Z.; Rush, A.; Charney, D.; Wilson, A.F.; Sorant, A.J.M.; Papanicolaou, G.J.; Fava, M.; Trivedi, M.H.; Wisniewski, S.R.; Laje, G.; et al. Association Between a Functional Serotonin Transporter Promoter Polymorphism and Citalopram Treatment in Adult Outpatients With Major Depression. Arch. Gen. Psychiatry 2007, 64, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, D.; Gonda, X.; Petschner, P.; Edes, A.; Eszlari, N.; Bagdy, G.; Juhasz, G. Antidepressant treatment response is modulated by genetic and environmental factors and their interactions. Ann. Gen. Psychiatry 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelli, L.; Marino, E.; Pirovano, A.; Calati, R.; Zanardi, R.; Colombo, C.; Serretti, A. Interaction between SERTPR and stressful life events on response to antidepressant treatment. Eur. Neuropsychopharmacol. 2009, 19, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Keers, R.R.; Uher, R.; Huezo-Diaz, P.; Smith, R.P.; Jaffee, S.S.; Rietschel, M.; Henigsberg, N.N.; Kozel, D.; Mors, O.; Maier, W.W.; et al. Interaction between serotonin transporter gene variants and life events predicts response to antidepressants in the GENDEP project. Pharmacogenomics J. 2010, 11, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philibert, R.; Madan, A.; Andersen, A.; Cadoret, R.; Packer, H.; Sandhu, H. Serotonin transporter mRNA levels are associated with the methylation of an upstream CpG island. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2006, 101–105. [Google Scholar] [CrossRef]
- Philibert, R.A.; Sandhu, H.; Hollenbeck, N.; Gunter, T.; Adams, W.; Madan, A. The relationship of5HTT(SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2007, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnally, E.L.; Capitanio, J.P.; Leibel, R.; Deng, L.; LeDuc, C.; Haghighi, F.; Mann, J.J. Epigenetic regulation of serotonin transporter expression and behavior in infant rhesus macaques. Genes Brain Behav. 2010, 9, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ijzendoorn, M.H.; Caspers, K.; Bakermans-Kranenburg, M.J.; Beach, S.R.H.; Philibert, R.A. Methylation Matters: Interaction Between Methylation Density and Serotonin Transporter Genotype Predicts Unresolved Loss or Trauma. Biol. Psychiatry 2010, 68, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Alasaari, J.S.; Lagus, M.; Ollila, H.M.; Toivola, A.; Kivimäki, M.; Vahtera, J.; Kronholm, E.; Härmä, M.; Puttonen, S.; Paunio, T. Environmental Stress Affects DNA Methylation of a CpG Rich Promoter Region of Serotonin Transporter Gene in a Nurse Cohort. PLoS ONE 2012, 7, e45813. [Google Scholar] [CrossRef] [PubMed]
- Duman, E.A.; Canli, T. Influence of life stress, 5-HTTLPR genotype, and SLC6A4 methylation on gene expression and stress response in healthy Caucasian males. Biol. Mood Anxiety Disord. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Alexander, N.; Wankerl, M.; Hennig, J.; Miller, R.; Zänkert, S.; Steudte-Schmiedgen, S.; Stalder, T.; Kirschbaum, C. DNA methylation profiles within the serotonin transporter gene moderate the association of 5-HTTLPR and cortisol stress reactivity. Transl. Psychiatry 2014, 4, e443. [Google Scholar] [CrossRef] [Green Version]
- Frodl, T.; Szyf, M.; Carballedo, A.; Ly, V.; Dymov, S.; Vaisheva, F.; Morris, D.; Fahey, C.; Meaney, J.F.; Gill, M.; et al. DNA methylation of the serotonin transporter gene (SLC6A4) is associated with brain function involved in processing emotional stimuli. J. Psychiatry Neurosci. 2015, 40, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Ur, E.; White, P.D.; Grossman, A. Hypothesis: Cytokines may be activated to cause depressive illness and chronic fatigue syndrome. Eur. Arch. Psychiatry Clin. Neurosci. 1992, 241, 317–322. [Google Scholar] [CrossRef]
- Wichers, M.; Maes, M. The psychoneuroimmuno-pathophysiology of cytokine-induced depression in humans. Int. J. Neuropsychopharmacol. 2002, 5, 375–388. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Boorman, E.; Romano, G.F.; Russell, A.; Mondelli, V.; Pariante, C.M. Are Mood and Anxiety Disorders Inflammatory Diseases? Psychiatr. Ann. 2015, 45, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Von Känel, R.; Hepp, U.; Kraemer, B.; Traber, R.; Keel, M.; Mica, L.; Schnyder, U. Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J. Psychiatr. Res. 2007, 41, 744–752. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Gałecki, P.; Talarowska, M. Inflammatory theory of depression. Psychiatr. Polska 2018, 52, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Halaris, A. Inflammation and depression but where does the inflammation come from? Curr. Opin. Psychiatry 2019, 32, 422–428. [Google Scholar] [CrossRef]
- Halaris, A.; Leonard, B.E. Unraveling the complex interplay of immunometabolic systems that contribute to the neuropro-gression of psychiatric disorders. Neurol. Psychiatry Brain Res. 2019, 32, 111–121. [Google Scholar] [CrossRef]
- Topić, R.; Miličić, D.; Štimac, Z.; Lončar, M.; Velagić, V.; Marčinko, D.; Jakovljević, M. Somatic comorbidity, metabolic syndrome, cardiovascular risk, and CRP in patients with recurrent depressive disorders. Croat. Med. J. 2013, 54, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef]
- Vashist, S.K.; Venkatesh, A.; Schneider, E.M.; Beaudoin, C.; Luppa, P.B.; Luong, J.H. Bioanalytical advances in assays for C-reactive protein. Biotechnol. Adv. 2016, 34, 272–290. [Google Scholar] [CrossRef]
- Jeon, S.W.; Kim, Y.-K. Inflammation-induced depression: Its pathophysiology and therapeutic implications. J. Neuroimmunol. 2017, 313, 92–98. [Google Scholar] [CrossRef]
- Lang, U.E.; Borgwardt, S. Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef]
- Wium-Andersen, M.K.; Ørsted, D.D.; Nordestgaard, B.G. Elevated C-Reactive Protein, Depression, Somatic Diseases, and All-Cause Mortality: A Mendelian Randomization Study. Biol. Psychiatry 2014, 76, 249–257. [Google Scholar] [CrossRef]
- Osimo, E.F.; Baxter, L.J.; Lewis, G.; Jones, P.B.; Khandaker, G.M. Prevalence of low-grade inflammation in depression: A systematic review and meta-analysis of CRP levels. Psychol. Med. 2019, 49, 1958–1970. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of Depression With C-Reactive Protein, IL-1, and IL-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haapakoski, R.; Mathieu, J.; Ebmeier, K.P.; Alenius, H.; Kivimäki, M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015, 49, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Dargél, A.A.; Godin, O.; Kapczinski, F.; Kupfer, D.J.; Leboyer, M. C-Reactive Protein Alterations in Bipolar Disorder: A Meta–Analysis. J. Clin. Psychiatry 2015, 76, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, B.S.; Steiner, J.; Bernstein, H.-G.; Dodd, S.; Pasco, J.A.; Dean, O.; Nardin, P.; Gonçalves, C.; Berk, M. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: Meta-analysis and implications. Mol. Psychiatry 2015, 21, 554–564. [Google Scholar] [CrossRef]
- Moriarity, D.P.; Horn, S.R.; Kautz, M.M.; Haslbeck, J.M.; Alloy, L.B. How handling extreme C-reactive protein (CRP) values and regularization influences CRP and depression criteria associations in network analyses. Brain Behav. Immun. 2021, 91, 393–403. [Google Scholar] [CrossRef]
- Turkheimer, F.E.; Althubaity, N.; Schubert, J.; Nettis, M.A.; Cousins, O.; Dima, D.; Mondelli, V.; Bullmore, E.T.; Pariante, C.; Veronese, M. Increased serum peripheral C-reactive protein is associated with reduced brain barriers permeability of TSPO radioligands in healthy volunteers and depressed patients: Implications for inflammation and depression. Brain Behav. Immun. 2021, 91, 487–497. [Google Scholar] [CrossRef]
- Ramani, T.; Auletta, C.S.; Weinstock, D.; Mounho-Zamora, B.; Ryan, P.C.; Salcedo, T.W.; Bannish, G. Cytokines: The Good, the Bad, and the Deadly. Int. J. Toxicol. 2015, 34, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Schlichtiger, J.; Pekcec, A.; Bartmann, H.; Winter, P.; Fuest, C.; Soerensen, J.; Potschka, H. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br. J. Pharmacol. 2010, 160, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Gałecki, P.; Talarowska, M.; Bobińska, K.; Szemraj, J. COX-2 gene expression is correlated with cognitive function in recurrent depressive disorder. Psychiatry Res. 2014, 215, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Johansson, D.; Falk, A.; Marcus, M.M.; Svensson, T.H. Celecoxib enhances the effect of reboxetine and fluoxetine on cortical noradrenaline and serotonin output in the rat. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 39, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.I. Antiinflammatory and neuroprotective actions of COX2 inhibitors in the injured brain. Brain Behav. Immun. 2008, 22, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, O.; Krogh, J.; Mors, O.; Benros, M.E. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casolini, P.; Catalani, A.; Zuena, A.R.; Angelucci, L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res. 2002, 68, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Mendlewicz, J.; Kriwin, P.; Oswald, P.; Souery, D.; Alboni, S.; Brunello, N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: A pilot open-label study. Int. Clin. Psychopharmacol. 2006, 21, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef]
- Müller, N. Immunomodulation as Therapeutic Approach in Schizophrenia and Depression: State of the Art. In Immunology and Psychiatry: Current Topics in Neurotoxicity; Müller, N., Myint, A.M., Schwarz, M., Eds.; Springer: Cham, Switzerland, 2015; pp. 351–369. ISBN 978-3-319-13601-1. [Google Scholar]
- Akhondzadeh, S.; Tabatabaee, M.; Amini, H.; Abhari, S.A.A.; Abbasi, S.H.; Behnam, B. Celecoxib as adjunctive therapy in schizophrenia: A double-blind, randomized and placebo-controlled trial. Schizophr. Res. 2007, 90, 179–185. [Google Scholar] [CrossRef]
- Abbasi, S.-H.; Hosseini, F.; Modabbernia, A.; Ashrafi, M.; Akhondzadeh, S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: Randomized double-blind placebo-controlled study. J. Affect. Disord. 2012, 141, 308–314. [Google Scholar] [CrossRef]
- Halaris, A.; Cantos, A.; Johnson, K.; Hakimi, M.; Sinacore, J. Modulation of the inflammatory response benefits treatment-resistant bipolar depression: A randomized clinical trial. J. Affect. Disord. 2020, 261, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Edberg, D.; Hoppensteadt, D.; Walborn, A.; Fareed, J.; Sinacore, J.; Halaris, A. Plasma C-reactive protein levels in bipolar depression during cyclooxygenase-2 inhibitor combination treatment. J. Psychiatr. Res. 2018, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shirayama, Y.; Muneoka, K.; Suzuki, M.; Sato, K.; Hashimoto, K. Personality Traits as Risk Factors for Treatment-Resistant Depression. PLoS ONE 2013, 8, e63756. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shirayama, Y.; Muneoka, K.; Suzuki, M.; Sato, K.; Hashimoto, K. Low Openness on the Revised NEO Personality Inventory as a Risk Factor for Treatment-Resistant Depression. PLoS ONE 2013, 8, e71964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, R.T. Personality Pathology and Treatment Outcome in Major Depression: A Review. Am. J. Psychiatry 2002, 159, 359–371. [Google Scholar] [CrossRef]
- Gorwood, P.; Rouillon, F.; Even, C.; Falissard, B.; Corruble, E.; Moran, P. Treatment response in major depression: Effects of personality dysfunction and prior depression. Br. J. Psychiatry 2010, 196, 139–142. [Google Scholar] [CrossRef]
- Skodol, A.E.; Grilo, C.M.; Keyes, K.M.; Geier, T.; Grant, B.F.; Hasin, D.S. Relationship of Personality Disorders to the Course of Major Depressive Disorder in a Nationally Representative Sample. Am. J. Psychiatry 2011, 168, 257–264. [Google Scholar] [CrossRef]
- Souery, D.; Papakostas, G.I.; Trivedi, M.H. Treatment-resistant depression. J. Clin. Psychiatry 2006, 67 (Suppl. 6), 16–22. [Google Scholar]
- Duggan, C.F.; Lee, A.S.; Murray, R.M. Does Personality Predict Long-Term Outcome in Depression? Br. J. Psychiatry 1990, 157, 19–24. [Google Scholar] [CrossRef]
- Kornstein, S.G.; Schneider, R.K. Clinical features of treatment-resistant depression. J. Clin. Psychiatry 2001, 62 (Suppl. 1), 18–25. [Google Scholar] [PubMed]
- Klein, D.N.; Kotov, R.; Bufferd, S.J. Personality and Depression: Explanatory Models and Review of the Evidence. Annu. Rev. Clin. Psychol. 2011, 7, 269–295. [Google Scholar] [CrossRef] [Green Version]
- Ørstavik, R.E.; Kendler, K.S.; Czajkowski, N.; Tambs, K.; Reichborn-Kjennerud, T. The Relationship between Depressive Personality Disorder and Major Depressive Disorder: A Population-Based Twin Study. Am. J. Psychiatry 2007, 164, 1866–1872. [Google Scholar] [CrossRef]
- Wiles, N.; Thomas, L.; Abel, A.; Ridgway, N.; Turner, N.; Campbell, J.; Garland, A.; Hollinghurst, S.; Jerrom, B.; Kessler, D.; et al. Cognitive behavioural therapy as an adjunct to pharmacotherapy for primary care based patients with treatment resistant depression: Results of the CoBalT randomised controlled trial. Lancet 2013, 381, 375–384. [Google Scholar] [CrossRef]
- Keller, M.B.; McCullough, J.P.; Klein, D.N.; Arnow, B.; Dunner, D.L.; Gelenberg, A.J.; Markowitz, J.C.; Nemeroff, C.B.; Russell, J.M.; Thase, M.E.; et al. A Comparison of Nefazodone, the Cognitive Behavioral-Analysis System of Psychotherapy, and Their Combination for the Treatment of Chronic Depression. N. Engl. J. Med. 2000, 342, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocsis, J.H.; Gelenberg, A.J.; Rothbaum, B.O.; Klein, D.N.; Trivedi, M.H.; Manber, R.; Keller, M.B.; Leon, A.C.; Wisniewski, S.R.; Arnow, B.A.; et al. Cognitive Behavioral Analysis System of Psychotherapy and Brief Supportive Psychotherapy for Augmentation of Antidepressant Nonresponse in Chronic DepressionThe REVAMP TrialAugmentation With CBASP and BSP for Depression. Arch. Gen. Psychiatry 2009, 66, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thase, M.E.; Friedman, E.S.; Biggs, M.M.; Wisniewski, S.R.; Trivedi, M.H.; Luther, J.F.; Fava, M.; Nierenberg, A.A.; McGrath, P.J.; Warden, D.; et al. Cognitive Therapy Versus Medication in Augmentation and Switch Strategies as Second-Step Treatments: A STAR*D Report. Am. J. Psychiatry 2007, 164, 739–752. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halaris, A.; Sohl, E.; Whitham, E.A. Treatment-Resistant Depression Revisited: A Glimmer of Hope. J. Pers. Med. 2021, 11, 155. https://doi.org/10.3390/jpm11020155
Halaris A, Sohl E, Whitham EA. Treatment-Resistant Depression Revisited: A Glimmer of Hope. Journal of Personalized Medicine. 2021; 11(2):155. https://doi.org/10.3390/jpm11020155
Chicago/Turabian StyleHalaris, Angelos, Emilie Sohl, and Elizabeth A. Whitham. 2021. "Treatment-Resistant Depression Revisited: A Glimmer of Hope" Journal of Personalized Medicine 11, no. 2: 155. https://doi.org/10.3390/jpm11020155
APA StyleHalaris, A., Sohl, E., & Whitham, E. A. (2021). Treatment-Resistant Depression Revisited: A Glimmer of Hope. Journal of Personalized Medicine, 11(2), 155. https://doi.org/10.3390/jpm11020155