Modified Significance Analysis of Microarrays in Heterogeneous Diseases


Abstract
:1. Background
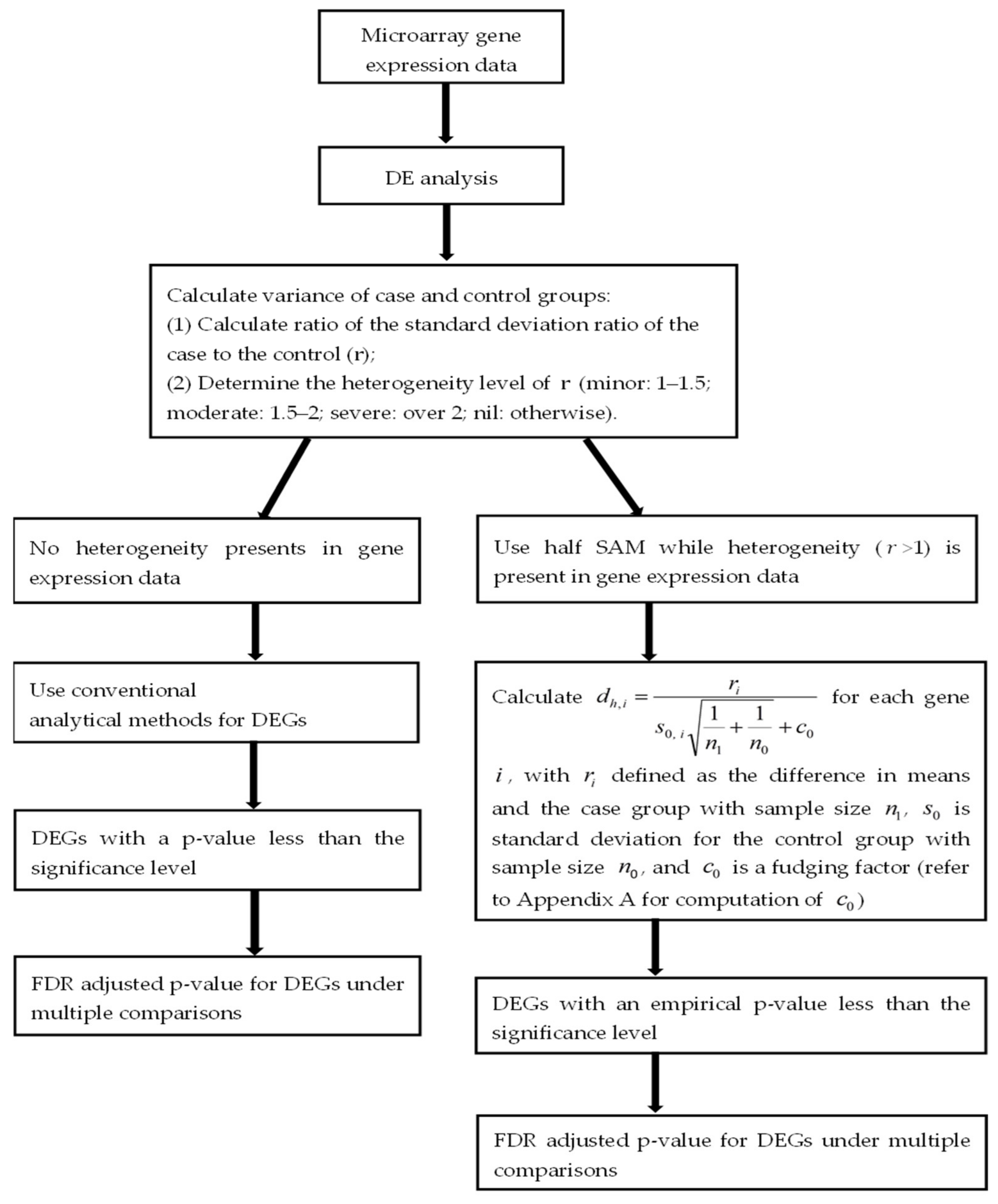
2. Materials and Methods
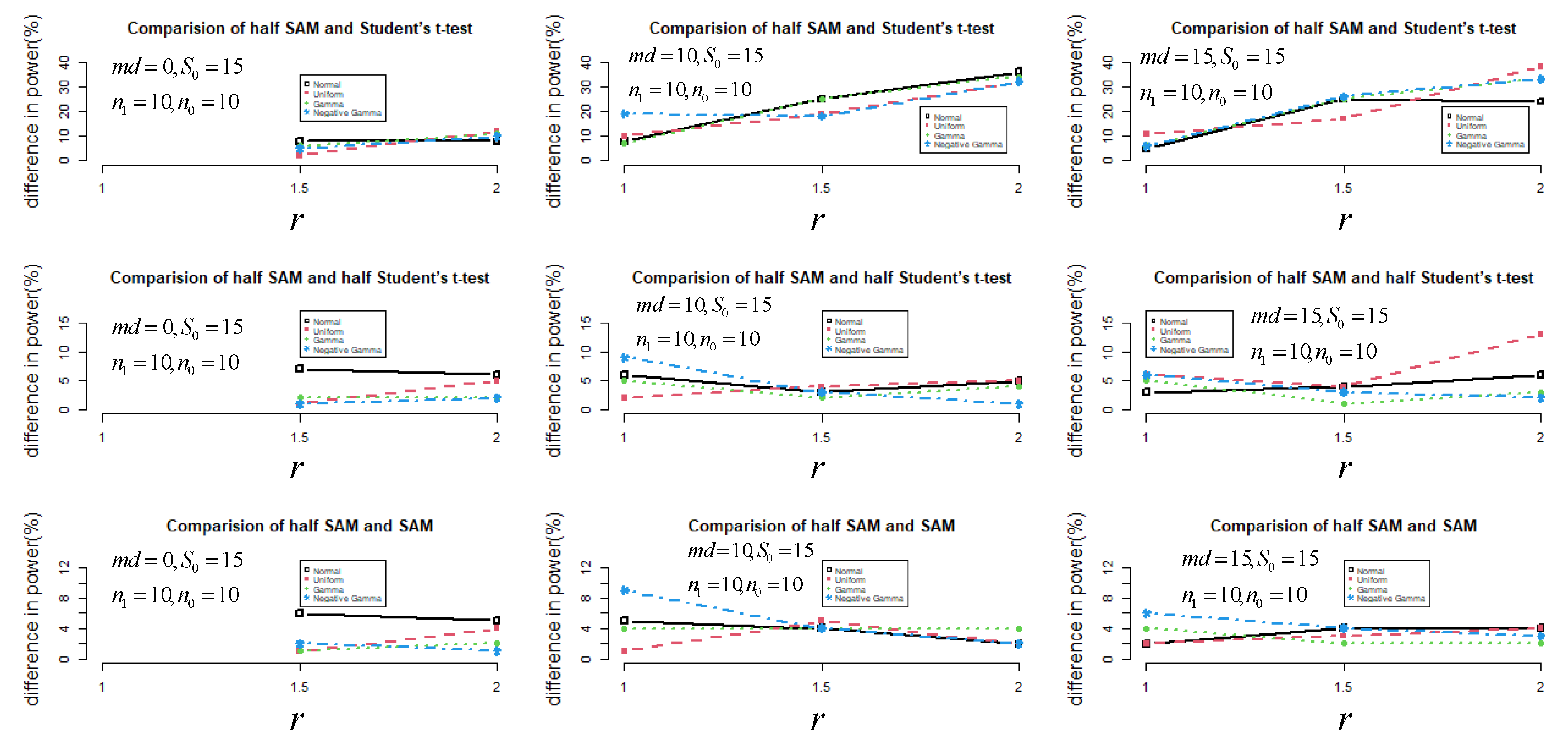
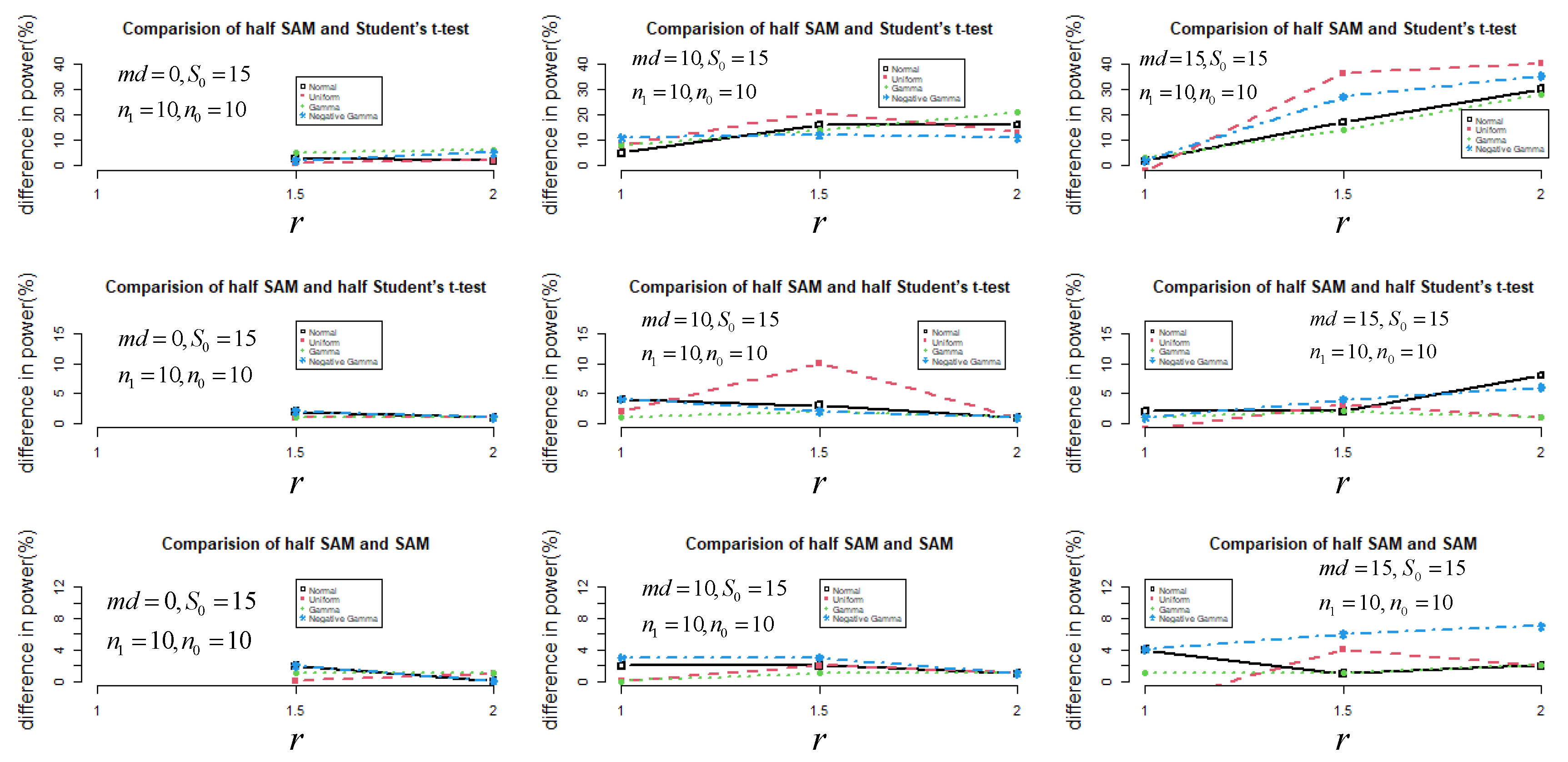
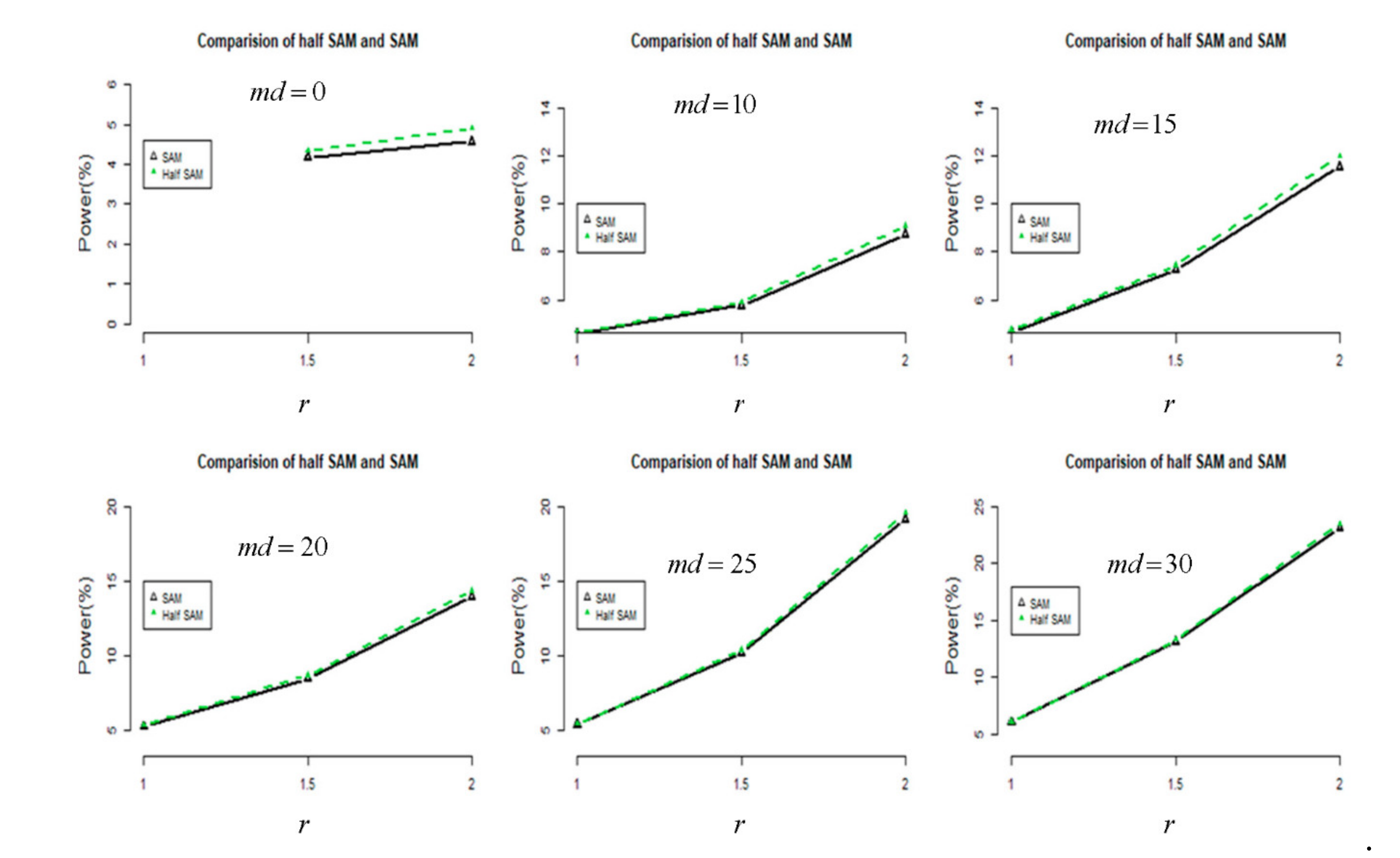
2.1. Monte Carlo Simulation
2.2. An Example for Demonstration
3. Results
3.1. Simulation Results
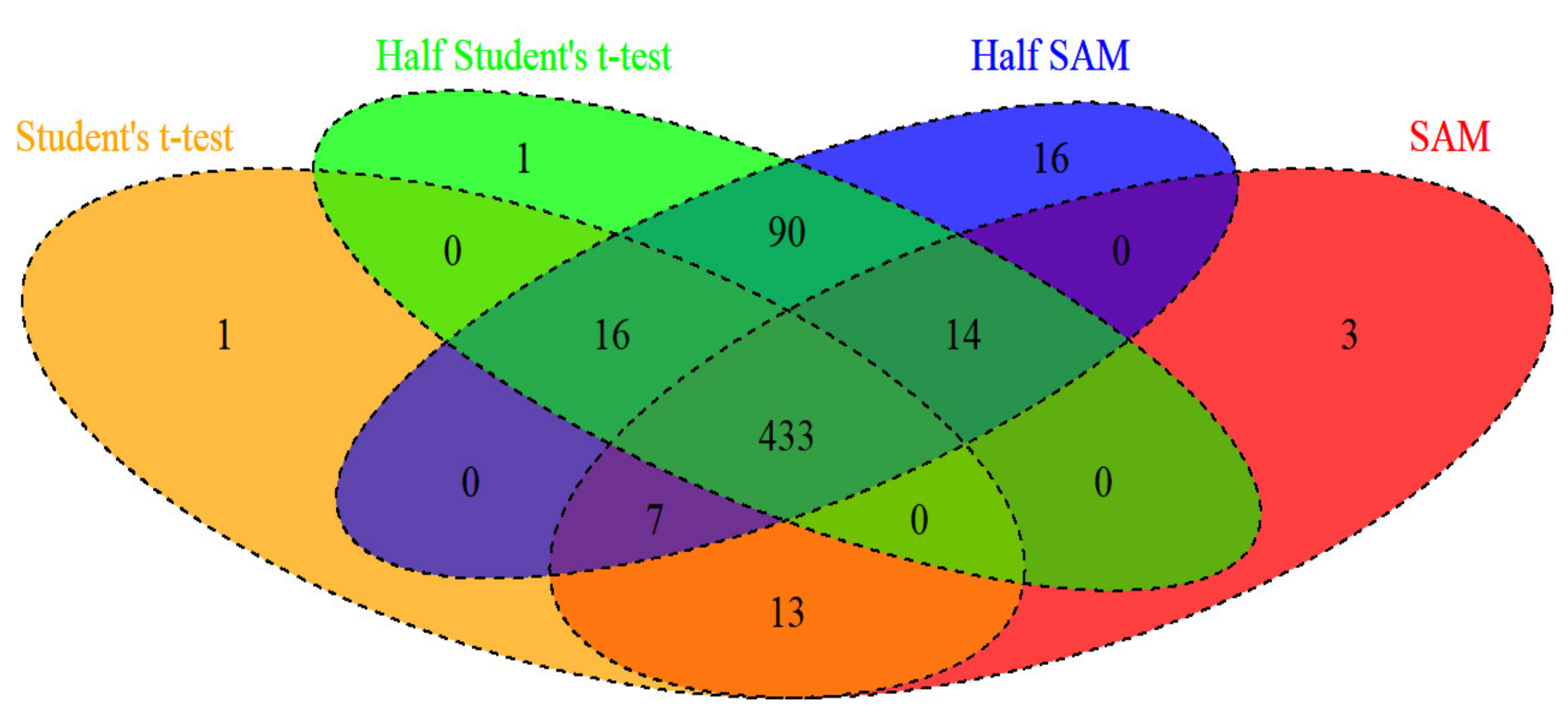
3.2. Main Results for Colon Cancer Data
3.3. Main Results for RNA-Seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Ethics Approval and Consent to Participate
Conflicts of Interest
Appendix A
- Let and let be the th percentile of the values.
- Compute for (0, 0.01, 0.02, …, 1.0) for the 100 quantiles, in turn, of the values denoted by < < … < .
- Consider (0, 0.05, 0.10, …, 1.0) for the 20 quantiles in turn.
- (1)
- Let = mad(| [, )) divided by 0.64, where mad is defined as the median absolute deviation from the median.
- (2)
- Let the coefficient of variation of the values be denoted as .
- Determine according to certain via the criterion = argmin []. The fudging factor is determined by the value in the end.
References
- Brown, P.O.; Botstein, D. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 1999, 21, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S. Array of hope. Nat. Genet. 1999, 21, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nat. Cell Biol. 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Tran-script assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differen-tiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Troyanskaya, O.G.; Garber, M.E.; Brown, P.O.; Botstein, D.; Altman, R.B. Nonparametric methods for identifying differen-tially expressed genes in microarray data. Bioinformatics 2002, 18, 1454–1561. [Google Scholar] [CrossRef]
- Tzeng, I.-S.; Chen, L.-S.; Chang, S.; Lee, Y.-L.L.; Bahler, J. Detecting differentially expressed genes of heterogeneous and positively skewed data using half Johnson’s modified t-test. Cogent Biol. 2016, 2, 1220066. [Google Scholar] [CrossRef]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differ-entially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Smyth, G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.A.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Tzeng, I.-S.; Lee, W.-C. Detecting differentially expressed genes in heterogeneous diseases using control-only analysis of variance. Ann. Epidemiol. 2012, 22, 598–602. [Google Scholar] [CrossRef]
- Yeoh, E.-J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Thieblemont, C.; Mayer, A.; Dumontet, C.; Barbier, Y.; Callet-Bauchu, E.; Felman, P.; Berger, F.; Ducottet, X.; Martin, C.; Salles, G.; et al. Primary thyroid lymphoma is a heterogeneous disease. J. Clin. Endocrinol. Metab. 2002, 87, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, K.V.D.; Govaerts, P.; De Leenheer, E.; Schatteman, I.; Verstreken, M.; Chen, W.; Declau, F.; Cremers, C.; Van De Heyning, P.; Offeciers, F.; et al. Otosclerosis: A genetically heterogeneous disease involving at least three different genes. Bone 2002, 30, 624–630. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; Wang, X.; Medema, J.P.; Vermeulen, L. Colorectal Cancer Heterogeneity and Targeted Therapy: A Case for Molecular Disease Subtypes. Cancer Res. 2015, 75, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-L.; Lee, W.-C. Detecting differentially expressed genes in heterogeneous diseases using half Student’s t-test. Int. J. Epidemiol. 2010, 39, 1597–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudoit, S.; Yang, Y.H.; Callow, M.J.; Speed, T.P. Statistical methods for identifying differentially expressed genes in repli-cated cDNA microarray experiments. Stat. Sin. 2002, 12, 111–139. [Google Scholar]
- Alon, U.; Barkai, N.; Notterman, D.A.; Gish, K.; Ybarra, S.; Mack, D.; Levine, A.J. Broad patterns of gene expression re-vealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1999, 96, 6745–6750. [Google Scholar] [CrossRef] [Green Version]
- Giles, P.J.; Kipling, D. Normality of oligonucleotide microarray data and implications for parametric statistical analyses. Bioinformatics 2003, 19, 2254–2262. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 10 November 2020).
- Efron, B.; Tibshirani, R. An Introduction to the Bootstrap; Chapman & Hall: New York, NY, USA, 1993. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Himes, B.E.; Jiang, X.; Wagner, P.; Hu, R.; Wang, Q.; Klanderman, B.; Whitaker, R.M.; Duan, Q.; Lasky-Su, J.; Nikolos, C.; et al. RNA-Seq Transcriptome Profiling Identifies CRISPLD2 as a Glucocorticoid Responsive Gene that Modulates Cytokine Function in Airway Smooth Muscle Cells. PLoS ONE 2014, 9, e99625. [Google Scholar] [CrossRef]
- Le, C.T.; Pan, W.; Lin, J. A mixture model approach to detecting differentially expressed genes with microarray data. Funct. Integr. Genom. 2003, 3, 117–124. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R.; Storey, J.D.; Tusher, V. Empirical Bayes Analysis of a Microarray Experiment. J. Am. Stat. Assoc. 2001, 96, 1151–1160. [Google Scholar] [CrossRef]
- Li, J.; Tibshirani, R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-Seq data. Stat. Methods Med Res. 2011, 22, 519–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xi, Y.; Sung, S.; Qiao, H. RNA-seq assistant: Machine learning based methods to identify more transcriptional regulated genes. BMC Genom. 2018, 19, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roobaert, D.; Karakoulas, G.; Chawla, N.V. Information Gain, Correlation and Support Vector Machines. Comput. Intell. 2008, 207, 463–470. [Google Scholar] [CrossRef]
- Kerr, M.K.; Churchill, G.A. Experimental design for gene expression microarrays. Biostatistics 2001, 2, 183–201. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normaliza-tion, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wong, W.H. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 2001, 98, 31–36. [Google Scholar] [CrossRef]
- Kadota, K.; Nakai, Y.; Shimizu, K. Ranking differentially expressed genes from Affymetrix gene expression data: Methods with reproducibility, sensitivity, and specificity. Algorithms Mol. Biol. 2009, 4, 7. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Power % (Type I Error) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t-Test | Half t-Test | SAM | Half SAM | t-Test | Half t-Test | SAM | Half SAM | t-Test | Half t-Test | SAM | Half SAM | |
| Normal distribution | ||||||||||||
| 1 | (0.02) | (0.05) | (0.04) | (0.05) | 28 | 30 | 31 | 36 | 60 | 62 | 63 | 65 |
| 1.5 | 4 | 5 | 6 | 12 | 14 | 36 | 35 | 39 | 39 | 60 | 60 | 64 |
| 2 | 7 | 9 | 10 | 15 | 8 | 39 | 42 | 44 | 28 | 46 | 48 | 52 |
| Non-normal distribution | ||||||||||||
| 1 | (0.05) | (0.05) | (0.04) | (0.04) | 28 | 36 | 37 | 38 | 55 | 60 | 64 | 66 |
| 1.5 | 8 | 9 | 9 | 10 | 19 | 34 | 33 | 38 | 38 | 51 | 52 | 55 |
| 2 | 7 | 14 | 15 | 19 | 14 | 41 | 44 | 46 | 19 | 44 | 53 | 57 |
| Skew-to-right distribution | ||||||||||||
| 1 | (0.03) | (0.05) | (0.03) | (0.04) | 28 | 30 | 31 | 35 | 46 | 47 | 48 | 52 |
| 1.5 | 7 | 11 | 12 | 13 | 17 | 40 | 38 | 42 | 37 | 61 | 60 | 62 |
| 2 | 4 | 13 | 13 | 15 | 9 | 39 | 39 | 43 | 24 | 54 | 55 | 57 |
| Skew-to-left distribution | ||||||||||||
| 1 | (0.03) | (0.04) | (0.03) | (0.03) | 21 | 31 | 31 | 40 | 55 | 55 | 55 | 61 |
| 1.5 | 6 | 10 | 9 | 11 | 20 | 35 | 34 | 38 | 39 | 62 | 61 | 65 |
| 2 | 5 | 13 | 14 | 15 | 12 | 43 | 42 | 44 | 30 | 61 | 60 | 63 |
| Test Methods | ||||
|---|---|---|---|---|
| Student’s t-Test | SAM Score | Half Student’s t-Test | Half SAM Score | |
| Level of significance | ||||
| 0.05 | 470(23.50%) | 470(23.50%) | 554(27.70%) | 576(28.80%) |
| 0.01 | 239(11.95%) | 240(12.00%) | 300(15.00%) | 334(16.70%) |
| 0.005 | 179(8.95%) | 177(8.85%) | 250(12.50%) | 280(14.00%) |
| 0.001 | 74(3.70%) | 85(4.25%) | 141(7.05%) | 178(8.90%) |
| FDR | ||||
| 0.05 | 107(5.35%) | 119(5.95%) | 216(10.3%) | 265(13.25%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzeng, I.-S. Modified Significance Analysis of Microarrays in Heterogeneous Diseases. J. Pers. Med. 2021, 11, 62. https://doi.org/10.3390/jpm11020062
Tzeng I-S. Modified Significance Analysis of Microarrays in Heterogeneous Diseases. Journal of Personalized Medicine. 2021; 11(2):62. https://doi.org/10.3390/jpm11020062
Chicago/Turabian StyleTzeng, I-Shiang. 2021. "Modified Significance Analysis of Microarrays in Heterogeneous Diseases" Journal of Personalized Medicine 11, no. 2: 62. https://doi.org/10.3390/jpm11020062
APA StyleTzeng, I. -S. (2021). Modified Significance Analysis of Microarrays in Heterogeneous Diseases. Journal of Personalized Medicine, 11(2), 62. https://doi.org/10.3390/jpm11020062