
The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects

{kind=link}
{kind=link}
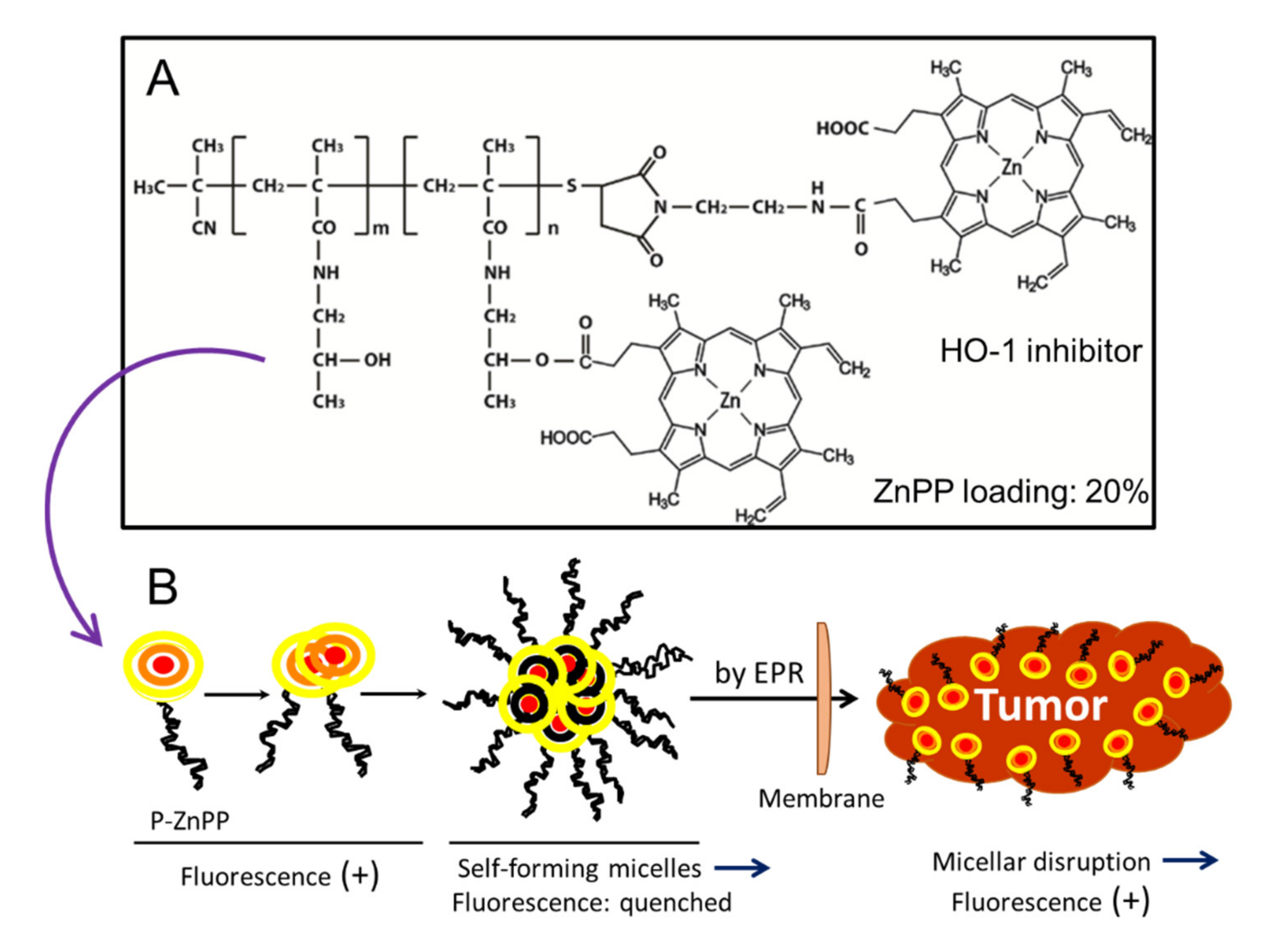{kind=link}
{kind=link}
{kind=link}
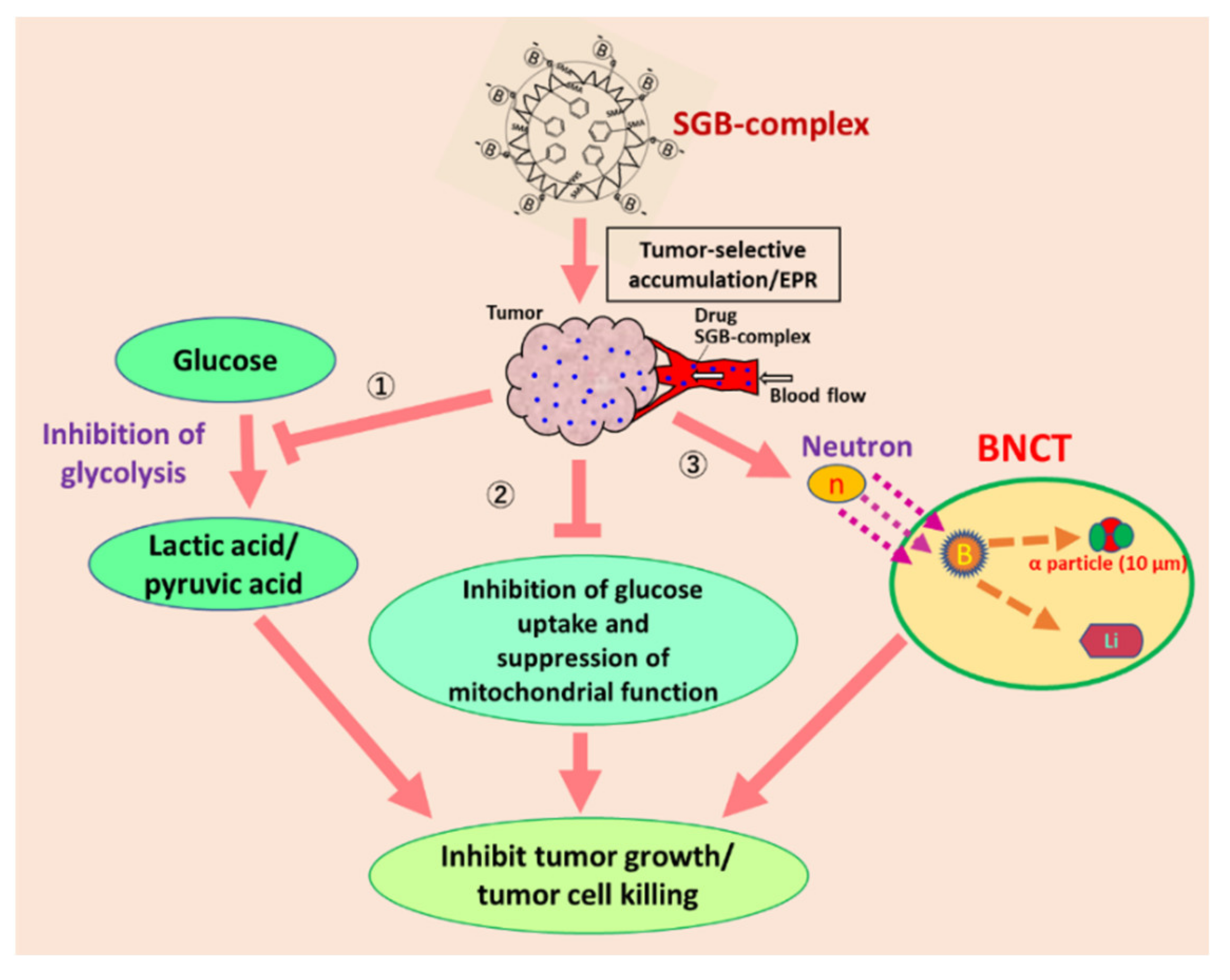{kind=link}
{kind=link}
Abstract
:1. Background: Discovery of the Enhanced Permeability and Retention (EPR) Effect, Criticism, and Reality
1.1. Status Quo of Enhanced Permeability and Retention (EPR) Effect and Tumor Targeting
1.2. Issue Concerns Passive Targeting to Tumor vs. the EPR Effect Driven Tumor Targeting
1.3. Inflammation and EPR Effect Observed in Bacterial Infection Protease and Permeability Inducing Factors; Bradykinin and Other Mediators
2. Nanomedicines: Proceeding from Tissue EPR Effects to Tumor Cellular Uptake to Molecular Targets in Tumor Cells
3. Future Prospects for the EPR Effect: Toward Clinical Application
3.1. Restoration of Tumor Blood Flow and Augmentation of the EPR Effect
3.2. Arterial Infusion of Nanomedicines with Extremely High Accumulation in Tumors
4. Enhancement of Cancer Chemotherapy, Utilization of Photodynamic Therapy (PDT), Innovation in Boron Neutron Capture Therapy (BNCT), and Use of Reactive Oxygen Species (ROS)/Reactive Nitrogen Species (RNS) as Scavengers for Cancer and Inflammation via Nanodrugs
4.1. Enhancement of Photodynamic Therapy (PDT)
4.2. A Hot Progress in Boron Neutron Capture Therapy (BNCT) with Boron Nanomedicines
5. Development of ROS and RNS Generators or Scavengers Utilizing the Advantages of Nanodrugs, and Future Clinical Applications
5.1. Elimination of Toxic Free Radical ROS/RNS in Infection and Cancer by Using Nanomedicines
5.2. Using ROS/RNS Generation to Kill Cancers by Means of ROS-Generating Polymer-Conjugated Enzymes, or Rescuing ROS-Caused Damage by Means of Enzyme Replacement Therapy via Conjugation with Synthetic Polymers
6. Concluding Remarks
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumortropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Noguchi, Y.; Wu, J.; Duncan, R.; Strohalm, J.; Ulbrich, K.; Akaike, T.; Maeda, H. Early phase tumor accumulation of macromolecules: A great difference in clearance rate between tumor and normal tissues. Jpn. J. Cancer Res. 1998, 89, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Maeda, H. The link between infection and cancer: Tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci. 2013, 104, 779–789. [Google Scholar] [CrossRef]
- Maeda, H. Polymer therapeutics and the EPR effect. J. Drug Target. 2017, 25, 781–785. [Google Scholar] [CrossRef]
- Maeda, H.; Tsukigawa, K.; Fang, J. A retrospective 30 years after discovery of the EPR effect of solid tumors: Treatment, imaging, and next-generation PDT—problems, solutions, prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T. Drug delivery research in Europe. J. Control. Release 2012, 161, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, J.P.; Das Neves, J.; De La Fuente, M.; Celia, C.; Florindo, H.; Günday-Türeli, N.; Popat, A.; Santos, J.L.; Sousa, F.; Schmid, R.; et al. The solid progress of nanomedicine. Drug Deliv. Transl. Res. 2020, 10, 726–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Park, K. The beginning of the end of the nanomedicine hype. J. Control. Release 2019, 305, 221–222. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nano-particle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin en-capsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Ding, Y.; Xu, Y.; Yang, W.; Niu, P.; Li, X.; Chen, Y.; Li, Z.; Liu, Y.; An, Y.; Liu, Y.; et al. Investigating the EPR effect of nanomedicines in human renal tumors via ex vivo perfusion strategy. Nano Today 2020, 35, 100970. [Google Scholar] [CrossRef]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; Lorusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Yamamoto, T.; Kamata, R.; Maeda, H. Pathogenesis of Serratial Infection: Activation of the Hageman Factor-Prekallikrein Cascade by Serratial Protease. J. Biochem. 1984, 96, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Kamata, R.; Yamamoto, T.; Matsumoto, K.; Maeda, H. A serratial protease causes vascular permeability reaction by activation of the Hageman factor-dependent pathway in guinea pigs. Infect. Immun. 1985, 48, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molla, A.; Yamamoto, T.; Akaike, T.; Miyoshi, S.; Maeda, H. Activation of Hageman factor and prekal-likrein and generation of kinin by various microbial proteinases. J. Biol. Chem. 1989, 264, 10589–10594. [Google Scholar] [CrossRef]
- Maruo, K.; Akaike, T.; Inada, Y.; Ohkubo, I.; Ono, T.; Maeda, H. Effect of microbial and mite proteases on low and high molecular weight kininogens. Generation of kinin and inactivation of thiol protease inhibitory activity. J. Biol. Chem. 1993, 268, 17711–17715. [Google Scholar] [CrossRef]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Okamoto, T.; Maruo, K.; Akaike, T. Kallikrein–kinin in infection and cancer. Immunopharmacology 1999, 43, 115–128. [Google Scholar] [CrossRef]
- Maeda, H.; Fang, J.; Inutsuka, T.; Kitamoto, Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int. Immunopharmacol. 2003, 3, 319–328. [Google Scholar] [CrossRef]
- Wu, J.; Akaike, T.; Maeda, H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998, 58, 159–165. [Google Scholar]
- Wu, J.; Akaike, T.; Hayashida, K.; Okamoto, T.; Okuyama, A.; Maeda, H. Enhanced vascular permea-bility in solid tumor involving peroxynitrite and matrix metalloproteinase. Jpn. J. Cancer Res. 2001, 92, 439–451. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y.; Kato, H. Purification and identification of [hydroxyprolyl3]bradykinin in ascitic fluid from a patient with gastric cancer. J. Biol. Chem. 1988, 263, 16051–16054. [Google Scholar] [CrossRef]
- Matsumura, Y.; Kimura, M.; Yamamoto, T.; Maeda, H. Involvement of the Kinin-generating Cascade in Enhanced Vascular Permeability in Tumor Tissue. Jpn. J. Cancer Res. 1988, 79, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maruo, K.; Kimura, M.; Yamamoto, T.; Konno, T.; Maeda, H. Kinin-generating Cascade in Advanced Cancer Patients andin vitroStudy. Jpn. J. Cancer Res. 1991, 82, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Maeda, H.; Iwai, K.; Tashiro, S.; Maki, S.; Morinaga, T.; Mochinaga, M.; Hiraoka, T.; Yokoyama, I. Effect of arterial administration of high-molecular-weight anticancer agent SMANCS with lipid lymphographic agent on hepatoma: A preliminary report. Eur. J. Cancer Clin. Oncol. 1983, 19, 1053–1065. [Google Scholar] [CrossRef]
- Maki, S.; Konno, T.; Maeda, H. Image enhancement in computerized tomography for sensitive diagnosis of liver cancer and semiquantitation of tumor selective drug targeting with oily contrast medium. Cancer 1985, 56, 751–757. [Google Scholar] [CrossRef]
- Konno, T.; Maeda, H.; Iwai, K.; Maki, S.; Tashiro, S.; Uchida, M.; Miyauchi, Y. Selective targeting of anti-cancer drug and simultaneous image enhancement in solid tumors by arterially administered lipid contrast medium. Cancer 1984, 54, 2367–2374. [Google Scholar] [CrossRef]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef]
- Nagamitsu, A.; Greish, K.; Maeda, H. Elevating Blood Pressure as a Strategy to Increase Tumor-targeted Delivery of Macromolecular Drug SMANCS: Cases of Advanced Solid Tumors. Jpn. J. Clin. Oncol. 2009, 39, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect (EPR) for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [Green Version]
- Laginha, K.M.; Verwoert, S.; Charrois, G.J.; Allen, T.M. Determination of Doxorubicin Levels in Whole Tumor and Tumor Nuclei in Murine Breast Cancer Tumors. Clin. Cancer Res. 2005, 11, 6944–6949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Koziolová, E.; Chytil, P.; Tsukigawa, K.; Fang, J.; Haratake, M.; Ulbrich, K.; Etrych, T.; Maeda, H. Pronounced Cellular Uptake of Pirarubicin versus That of Other Anthracyclines: Comparison of HPMA Copolymer Conjugates of Pirarubicin and Doxorubicin. Mol. Pharm. 2016, 13, 4106–4115. [Google Scholar] [CrossRef]
- Nakamura, H.; Koziolová, E.; Chytil, P.; Etrych, T.; Haratake, M.; Maeda, H. Superior Penetration and Cytotoxicity of HPMA Copolymer Conjugates of Pirarubicin in Tumor Cell Spheroid. Mol. Pharm. 2019, 16, 3452–3459. [Google Scholar] [CrossRef]
- Oda, T.; Sato, F.; Maeda, H. Facilitated internalization of neocarzinostatin and its lipophilic polymer conjugate, SMANCS, into cytosol in acidic pH. J. Natl. Cancer Inst. 1987, 79, 1205–1211. [Google Scholar]
- Oda, T.; Maeda, H. Binding to and internalization by cultured cells of neocarzinostatin and enhance-ment of its actions by conjugation with lipophilic styrene-maleic acid copolymer. Cancer Res. 1987, 47, 3206–3211. [Google Scholar] [PubMed]
- Maeda, H.; Islam, W. Overcoming barriers for tumor-targeted drug delivery: The power of macromo-lecular anticancer drugs with the EPR effect and the modulation of vascular physiology. In Polymer-Protein Conjugation: From PEGylation and Beyond; Pasut, G., Zalipsky, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 41–58. [Google Scholar]
- Miyamoto, Y.; Oda, T.; Maeda, H. Comparison of the cytotoxic effects of the high- and low-molecular-weight anticancer agents on multidrug-resistant Chinese hamster ovary cells in vitro. Cancer Res. 1990, 50, 1571–1575. [Google Scholar]
- Navi, B.B.; Reiner, A.S.; Kamel, H.; Iadecola, C.; Okin, P.M.; Tagawa, S.T.; Panageas, K.S.; DeAngelis, L.M. Arterial thromboembolic events preceding the diagnosis of cancer in older persons. Blood 2019, 133, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Fang, J.; Maeda, H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009, 100, 2426–2430. [Google Scholar] [CrossRef] [PubMed]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the enhanced permeability and retention effect with nitric oxide-generating agents improves the therapeutic effects of nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nittayacharn, P.; Yuan, H.-X.; Hernandez, C.; Bielecki, P.; Zhou, H.; Exner, A.A. Enhancing Tumor Drug Distribution with Ultrasound-Triggered Nanobubbles. J. Pharm. Sci. 2019, 108, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Hwang, J.; Lee, K.; Choi, Y.; Seo, Y.; Jeon, H.; Hong, J.W.; Choi, J. Anti-Tumor Drug-Loaded Oxygen Nanobubbles for the Degradation of HIF-1α and the Upregulation of Reactive Oxygen Species in Tumor Cells. Cancers 2019, 11, 1464. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Nakamura, H.; Etrych, T.; Chytil, P.; Ohkubo, M.; Fang, J.; Ulbrich, K.; Maeda, H. Two step mechanisms of tumor selective delivery of N-(2-hydroxypropyl)methacrylamide copolymer conjugated with pirarubicin via an acid-cleavable linkage. J. Control. Release 2014, 174, 81–87. [Google Scholar] [CrossRef]
- Dozono, H.; Yanazume, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2015, 11, 101–106. [Google Scholar] [CrossRef]
- Okuno, S. Birth of Anticancer Agent Without Adverse Effect. In Revolution in Cancer Treatment; (Book in Japanese. Interviews with treated patients of HPMA-polymer conjugated. Follow up by interview with P-THP treated many patients over several months to years); Bungei Shunju Sha: Tokyo, Japan, 2016; pp. 1–284. [Google Scholar]
- Iwai, K.; Maeda, H.; Konno, T. Use of oily contrast medium for selective drug targeting to tumor: Enhanced therapeutic effect and X-ray image. Cancer Res. 1984, 44, 2115–2121. [Google Scholar]
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: Advantages in cancer chemotherapy. Adv. Drug Deliv. Rev. 1991, 6, 181–202. [Google Scholar] [CrossRef]
- Seymour, L.W.; Olliff, S.P.; Poole, C.J.; De Takats, P.G.; Orme, R.; Ferry, D.R.; Maeda, H.; Konno, T.; Kerr, D.J. A novel dosage approach for evaluation of SMANCS [poly-(styrene-co-maleyl-half-n-butylate)-neocarzinostatin] in the treatment of primary hepatocellular carcinoma. Int. J. Oncol. 1998, 12, 1217–1240. [Google Scholar] [CrossRef]
- Fang, J.; Liao, L.; Yin, H.; Nakamura, H.; Subr, V.; Ulbrich, K.; Maeda, H. Photodynamic therapy and imaging based on tumor-targeted nanoprobe, polymer-conjugated zinc protoporphyrin. Futur. Sci. OA 2015, 1, fso4. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Fang, J.; Nakamura, H. Great expectations for innovative PDT and tumor imaging using fluorescing nanophotosensitizers that utilizes EPR effect. JSMI Rep. 2015, 9, 3–10. [Google Scholar]
- Fang, J.; Tsukigawa, K.; Liao, L.; Yin, H.; Eguchi, K.; Maeda, H. Styrene-maleic acid-copolymer conju-gated zinc protoporphyrin as a candidate drug for tumor-targeted therapy and imaging. J. Drug Target. 2016, 24, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Vladimir, Š.; Islam, W.; Islam, R.; Ulbrich, K.; Maeda, H. N-(2-Hydroxypropyl) methacrylamide polymer conjugated pyropheophorbide-a, a promising tumor-targeted theranostic probe for photody-namic therapy and imaging. Eur. J. Pharm. Biopharm. 2018, 130, 165–176. [Google Scholar] [CrossRef]
- Nakamura, H.; Liao, L.; Hitaka, Y.; Tsukigawa, K.; Subr, V.; Fang, J.; Ulbrich, K.; Maeda, H. Micelles of zinc protoporphyrin conjugated to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer for imaging and light-induced antitumor effects in vivo. J. Control. Release 2013, 165, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Sawa, T.; Akaike, T.; Akuta, T.; Sahoo, S.K.; Khaled, G.; Hamada, A.; Maeda, H. In vivo antitumor activity of pegylated zinc protoporphyrin: Targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003, 63, 3567–3574. [Google Scholar] [PubMed]
- Hadzijusufovic, E.; Rebuzzi, L.; Gleixner, K.V.; Ferenc, V.; Peter, B.; Kondo, R.; Gruze, A.; Kneidinger, M.; Krauth, M.-T.; Mayerhofer, M. Targeting of heat-shock protein 32/heme oxygenase-1 in canine mastocytoma cells is associated with reduced growth and induction of apoptosis. Exp. Hematol. 2008, 36, 1461–1470. [Google Scholar] [CrossRef]
- Mayerhofer, M.; Gleixner, K.V.; Mayerhofer, J.; Hoermann, G.; Jaeger, E.; Aichberger, K.J.; Ott, R.G.; Greish, K.; Nakamura, H.; Derdak, S.; et al. Targeting of heat shock protein 32 (Hsp32)/heme oxygenase-1 (HO-1) in leukemic cells in chronic myeloid leukemia: A novel approach to overcome resistance against imatinib. Blood 2008, 111, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Kneidinger, M.; Cerny-Reiterer, S.; Rülicke, T.; Willmann, M.; Gleixner, K.V.; Blatt, K.; Hörmann, G.; Peter, B.; Samorapoompichit, P.; et al. The Hsp32 inhibitors SMA-ZnPP and PEG-ZnPP exert major growth-inhibitory effects on CD34+/CD38+ and CD34+/CD38-AML progenitor cells. Curr. Cancer Drug Targets 2012, 12, 51–63. [Google Scholar] [CrossRef]
- Fang, J.; Greish, K.; Qin, H.; Liao, L.; Nakamura, H.; Takeya, M.; Maeda, H. HSP32 (HO-1) inhibitor, copoly(styrene-maleic acid)-zinc protoporphyrin IX, a water-soluble micelle as anticancer agent: In vitro and in vivo anticancer effect. Eur. J. Pharm. Biopharm. 2012, 81, 540–547. [Google Scholar] [CrossRef]
- Islam, W.; Matsumoto, Y.; Fang, J.; Harada, A.; Nidome, T.; Ono, K.; Tsutsuki, H.; Sawa, T.; Sakurai, K.; Fukumitsu, N.; et al. Polymer conjugated glucosamine complexed with boric acid shows tumor-selective accumulation and simultaneous inhibition of glycolysis. Biomaterials 2021, 269, 120631. [Google Scholar] [CrossRef]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influen-za-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2- generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaike, T.; Suga, M.; Maeda, H. Free radicals in viral pathogenesis: Molecular mechanisms involving superoxide and NO. Proc. Soc. Exp. Boil. Med. 1998, 217, 64–73. [Google Scholar] [CrossRef]
- Akaike, T.; Maeda, H. Nitric oxide and virus infection. Immunology 2000, 101, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J. Tackle the free radicals damage in COVID-19. Nitric Oxide 2020, 102, 39–41. [Google Scholar] [CrossRef]
- Akaike, T.; Fujii, S.; Kato, A.; Yoshitake, J.; Miyamoto, Y.; Sawa, T.; Okamoto, S.; Suga, M.; Asakawa, M.; Nagai, Y.; et al. Viral mutation accelerated by nitric oxide production during infection in vivo. FASEB J. 2000, 14, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Akaike, T.; Maeda, H. Role of Nitric Oxide in Pathogenesis of Herpes Simplex Virus Encephalitis in Rats. Virology 1999, 256, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Akaike, T.; Okamoto, S.; Sawa, T.; Yoshitake, J.; Tamura, F.; Ichimori, K.; Miyazaki, K.; Sasamoto, K.; Maeda, H. 8-Nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Yoshitake, J.; Akaike, T.; Akuta, T.; Tamura, F.; Ogura, T.; Esumi, H.; Maeda, H. Nitric Oxide as an Endogenous Mutagen for Sendai Virus without Antiviral Activity. J. Virol. 2004, 78, 8709–8719. [Google Scholar] [CrossRef] [Green Version]
- Shashni, B.; Nagasaki, Y. Newly Developed Self-Assembling Antioxidants as Potential Therapeutics for the Cancers. J. Pers. Med. 2021, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biol. Targets Ther. 2009, 3, 349–358. [Google Scholar]
- Kamisaki, Y.; Wada, H.; Yagura, T.; Matsushima, A.; Inada, Y. Reduction in immunogenicity and clearance rate of Escherichia coli L-asparaginase by modification with monomethoxypolyethylene glycol. J. Pharmacol. Exp. Ther. 1981, 216, 410–414. [Google Scholar]
- Kimura, M.; Matsumura, Y.; Miyauchi, Y.; Maeda, H. A New Tactic for the Treatment of Jaundice: An Injectable Polymer-Conjugated Bilirubin Oxidase. Exp. Biol. Med. 1988, 188, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Wu, J.; Akaike, T.; Maeda, H. Tumor-targeting chemotherapy by a xanthine oxidase-polymer conjugate that generates oxygen-free radicals in tumor tissue. Cancer Res. 2000, 60, 666–671. [Google Scholar] [PubMed]
- Fang, J.; Deng, D.; Nakamura, H.; Akuta, T.; Quin, H.; Iyer, A.; Greish, K.; Maeda, H. Oxystress in-ducing antitumor therapeutics via tumor-targeted delivery of PEG-conjugated D-amino acid oxidase. Int. J. Cancer 2008, 122, 1135–1144. [Google Scholar] [CrossRef]
- Nakamura, H.; Fang, J.; Maeda, H. Protective role of D-amino acid oxidase against bacterial infection. Infect. Immun. 2012, 80, 1546–1553. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Fang, J.; Mizukami, T.; Nunoi, H.; Maeda, H. Pegylated D-amino acid oxidase restores bactericidal activity of neutrophils in chronic granulomatous disease via hypochlorite. Exp. Biol. Med. 2012, 237, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Iyer, A.K.; Seki, T.; Nakamura, H.; Greish, K.; Maeda, H. SMA–copolymer conjugate of AHPP: A polymeric inhibitor of xanthine oxidase with potential antihypertensive effect. J. Control. Release 2009, 135, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, N.; Ghandehari, H. Harnessing Extracellular Matrix Biology for Tumor Drug Delivery. J. Pers. Med. 2021, 11, 88. [Google Scholar] [CrossRef]
- Meiab, T.; Shashnia, B.; Maeda, H.; Nagasakiade, Y. Fibrinolytic tissue plasminogen activator installed redox-active nanoparticles (t-PA@iRNP) for cancer therapy. Biomaterials 2020, 259, 120290. [Google Scholar] [CrossRef]
- Daruwalla, J.; Greish, K.; Malcontenti-Wilson, C.; Muralidharan, V.; Iyer, A.; Maeda, H.; Christophi, C. Styrene maleic acid-pirarubicin disrupts tumor microcirculation and enhances the permeability of col-orectal liver metastases. J. Vasc. Res. 2009, 46, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Daruwalla, J.; Nikfarjam, M.; Greish, K.; Malcontenti-WilsonI, C.; Muralidharan, V.; Christophi, C.; Maeda, H. In vitro and in vivo evaluation of tumor targeting styrene-maleic acid copolymer-pirarubicin micelles: Survival improvement and inhibition of liver metastases. Cancer Sci. 2010, 101, 1866–1874. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, H. The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. J. Pers. Med. 2021, 11, 229. https://doi.org/10.3390/jpm11030229
Maeda H. The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. Journal of Personalized Medicine. 2021; 11(3):229. https://doi.org/10.3390/jpm11030229
Chicago/Turabian StyleMaeda, Hiroshi. 2021. "The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects" Journal of Personalized Medicine 11, no. 3: 229. https://doi.org/10.3390/jpm11030229
APA StyleMaeda, H. (2021). The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. Journal of Personalized Medicine, 11(3), 229. https://doi.org/10.3390/jpm11030229