
Patient-Derived Cancer Organoids for Precision Oncology Treatment

 ,
,  , ,
, ,
Abstract
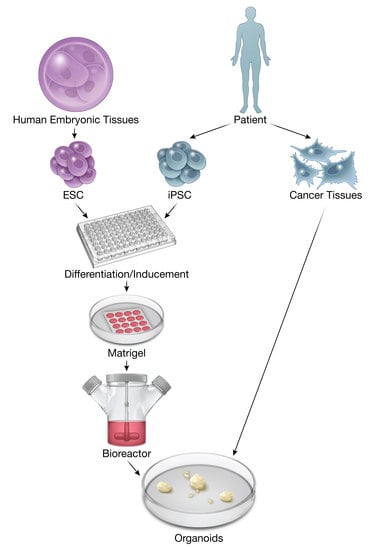
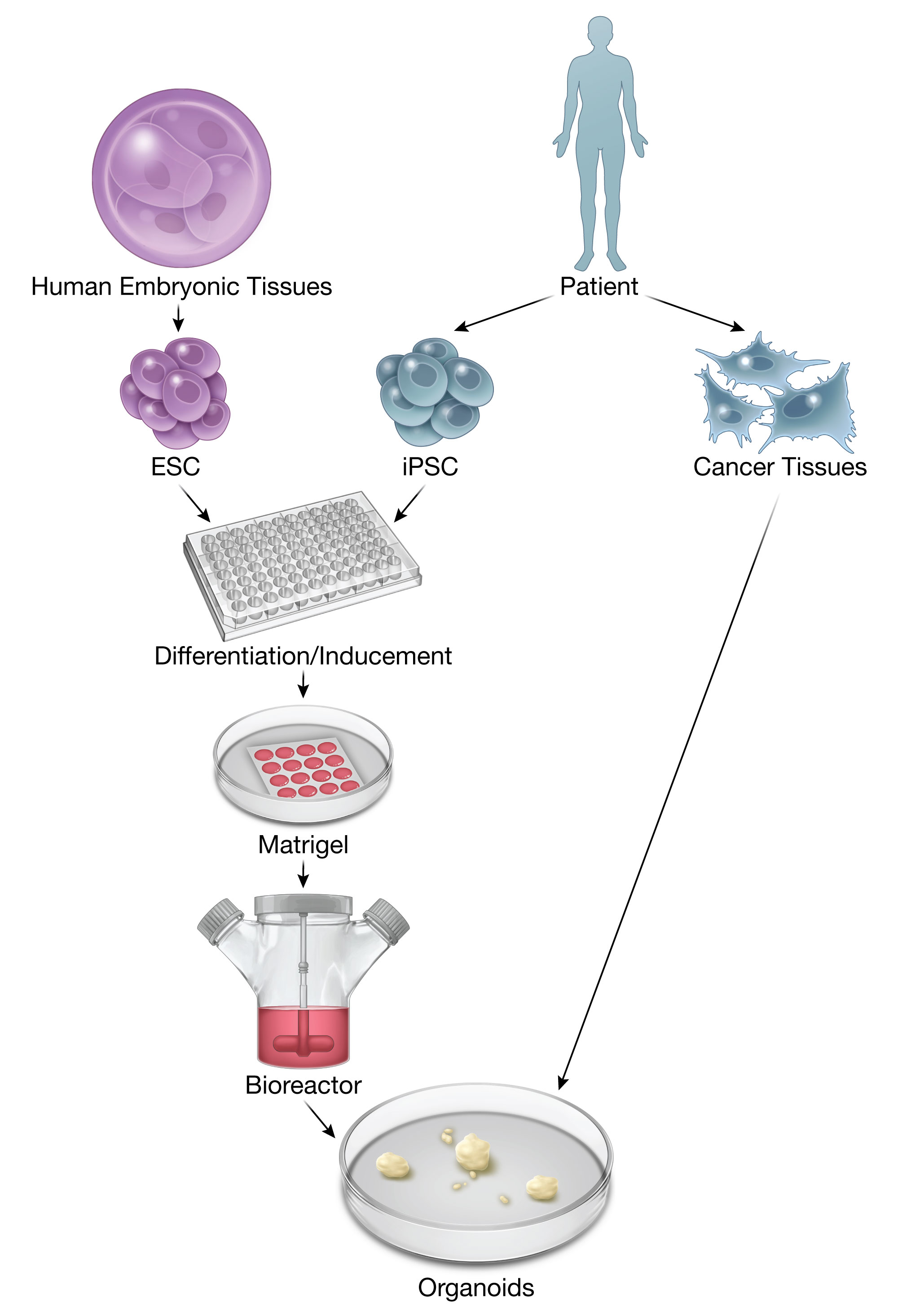
:
1. Introduction
2. History of Organoid Development
3. Current Models of Patient-Derived Cancer Glioma Organoids
4. Clinical Applications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. B 2017. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized medicine for patients with advanced cancer in the phase i program at MD Anderson: Validation and landmark analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef] [Green Version]
- Kamb, A. What’s wrong with our cancer models? Nat. Rev. Drug Discov. 2005. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Acker, T. Isolation and culture of primary glioblastoma cells from human tumor specimens. Methods Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Aboulkheyr Es, H.; Montazeri, L.; Aref, A.R.; Vosough, M.; Baharvand, H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018. [Google Scholar] [CrossRef]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, K.; Hjelmeland, A. Method for Efficient Transduction of Cancer Stem Cells. J. Cancer Stem Cell Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Feist, P.E.; Sidoli, S.; Liu, X.; Schroll, M.M.; Rahmy, S.; Fujiwara, R.; Garcia, B.A.; Hummon, A.B. Multicellular Tumor Spheroids Combined with Mass Spectrometric Histone Analysis to Evaluate Epigenetic Drugs. Anal. Chem. 2017, 89, 2773–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastomas. Pharmacol. Rev. 2018. [Google Scholar] [CrossRef] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Wang, M.; Wu, Z.; Li, N.; Zhang, J.; Yang, C. EZH2−, CHD4−, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.W.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009, 11, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Kijima, N.; Kanemura, Y. Mouse Models of Glioblastoma. Glioblastoma 2017. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020. [Google Scholar] [CrossRef]
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N.; Raghavan, V.; Li, Q.; Mer, A.S.; et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C., II; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017. [Google Scholar] [CrossRef]
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K.; Rowe, A.; Senthebane, D.A.; Almazyadi, M.A.M.; Patten, V.; Parker, M.I. Three-Dimensional Organoids in Cancer Research: The Search for the Holy Grail of Preclinical Cancer Modeling. Omics J. Integr. Biol. 2018. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, R.; Chiba, K.; Schaeffer, L.; Regalado, S.G.; Lai, C.S.; Gao, Q.; Kiani, S.; Farin, H.F.; Clevers, H.; Cost, G.J.; et al. Human intestinal tissue with adult stem cell properties derived from pluripotent stem cells. Stem Cell Rep. 2014. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Lewandowski, B.C.; Watson, J.; Aihara, E.; Iwatsuki, K.; Bachmanov, A.A.; Margolskee, R.F.; Jiang, P. Single Lgr5- or Lgr6-expressing taste stem/progenitor cells generate taste bud cells ex vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 16401–16406. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Little, M.H. Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 2016. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Kerjaschki, D.; Penninger, J.M. Generation of blood vessel organoids from human pluripotent stem cells. Nat. Protoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Yucer, N.; Holzapfel, M.; Jenkins Vogel, T.; Lenaeus, L.; Ornelas, L.; Laury, A.; Sareen, D.; Barrett, R.; Karlan, B.Y.; Svendsen, C.N. Directed Differentiation of Human Induced Pluripotent Stem Cells into Fallopian Tube Epithelium. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011. [Google Scholar] [CrossRef]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Van Gerwen, B.; Van Der Ven, M.; Van De Wetering, M.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. eLife 2016. [Google Scholar] [CrossRef] [Green Version]
- Weeber, F.; Van De Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-Van Hooijdonk, C.G.M.; Van Der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrò, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Translational applications of adult stem cell-derived organoids. Development 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.Z.; Han, R.R.; Qiu, G.Z.; Ju, X.C.; Lou, G.; Jin, W.L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; Van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Ming, G.L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020. [Google Scholar] [CrossRef]
- Golebiewska, A.; Hau, A.C.; Oudin, A.; Stieber, D.; Yabo, Y.A.; Baus, V.; Barthelemy, V.; Klein, E.; Bougnaud, S.; Keunen, O.; et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Nadauld, L.D.; Garcia, S.; Natsoulis, G.; Bell, J.M.; Miotke, L.; Hopmans, E.S.; Xu, H.; Pai, R.K.; Palm, C.; Regan, J.F.; et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluth, J.M.; Schackmann, R.C.J.; Gray, G.K.; Selfors, L.M.; Li, C.M.C.; Boedicker, M.; Kuiken, H.J.; Richardson, A.; Brock, J.; Garber, J.; et al. Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nat. Commun. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018. [Google Scholar] [CrossRef]
- Calandrini, C.; Schutgens, F.; Oka, R.; Margaritis, T.; Candelli, T.; Mathijsen, L.; Ammerlaan, C.; van Ineveld, R.L.; Derakhshan, S.; de Haan, S.; et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 2020. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Panoutsopoulos, A.A. Organoids, Assembloids, and Novel Biotechnology: Steps Forward in Developmental and Disease-Related Neuroscience. Neuroscientist 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Choi, S.; Kang, B.; Kong, J.H.; Kim, Y.; Yoon, W.H.; Lee, H.R.; Kim, S.E.; Kim, H.M.; Lee, H.S.; et al. Creation of bladder assembloids mimicking tissue regeneration and cancer. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolck, H.A.; Corrò, C.; Kahraman, A.; von Teichman, A.; Toussaint, N.C.; Kuipers, J.; Chiovaro, F.; Koelzer, V.H.; Pauli, C.; Moritz, W.; et al. Tracing Clonal Dynamics Reveals that Two- and Three-dimensional Patient-derived Cell Models Capture Tumor Heterogeneity of Clear Cell Renal Cell Carcinoma. Eur. Urol. Focus 2019. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.M.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014. [Google Scholar] [CrossRef] [Green Version]
- Seidlitz, T.; Merker, S.R.; Rothe, A.; Zakrzewski, F.; Von Neubeck, C.; Grützmann, K.; Sommer, U.; Schweitzer, C.; Schölch, S.; Uhlemann, H.; et al. Human gastric cancer modelling using organoids. Gut 2019, 68, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018, 174, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; Begora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, B.; Mathew, R.K.; Polson, E.S.; Williams, J.; Wurdak, H. Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020. [Google Scholar] [CrossRef]
- Degl’Innocenti, A.; di Leo, N.; Ciofani, G. Genetic Hallmarks and Heterogeneity of Glioblastoma in the Single-Cell Omics Era. Adv. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007. [Google Scholar] [CrossRef] [Green Version]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; Von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010. [Google Scholar] [CrossRef] [Green Version]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Blatt, V.; Pession, A.; Tallini, G.; Bertorelle, R.; Bartolini, S.; Calbucci, F.; Andreoli, A.; et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol. 2008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, G.A.; Oksdath, M.; Brown, M.P.; Ebert, L.M. New approaches to model glioblastoma in vitro using brain organoids: Implications for precision oncology. Transl. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- De Witte, C.J.; Espejo Valle-Inclan, J.; Hami, N.; Lõhmussaar, K.; Kopper, O.; Vreuls, C.P.H.; Jonges, G.N.; van Diest, P.; Nguyen, L.; Clevers, H.; et al. Patient-Derived Ovarian Cancer Organoids Mimic Clinical Response and Exhibit Heterogeneous Inter- and Intrapatient Drug Responses. Cell Rep. 2020. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Centenera, M.M.; Hickey, T.E.; Jindal, S.; Ryan, N.K.; Ravindranathan, P.; Mohammed, H.; Robinson, J.L.; Schiewer, M.J.; Ma, S.; Kapur, P.; et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Omuro, A.M.P.; Ravelo, A.; Sommer, N.; Guerin, A.; Ionescu-Ittu, R.; Shi, S.; Macalalad, A.; Uhm, J.H. Overall survival in patients with glioblastoma before and after bevacizumab approval. Curr. Med. Res. Opin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Fujii, T.; Yamamoto, T.; Takami, H.; Yoshioka, I.; Yamaki, S.; Sonohara, F.; Shibuya, K.; Motoi, F.; Hirano, S.; et al. Phase I/II study of adding intraperitoneal paclitaxel in patients with pancreatic cancer and peritoneal metastasis. Br. J. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Irtenkauf, S.M.; Sobiechowski, S.; Hasselbach, L.A.; Nelson, K.K.; Transou, A.D.; Carlton, E.T.; Mikkelsen, T.; De Carvalho, A.C. Optimization of glioblastoma mouse orthotopic xenograft models for translational research. Comp. Med. 2017, 67, 300–314. [Google Scholar] [PubMed]
- Peterson, J.K.; Houghton, P.J. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur. J. Cancer 2004, 40, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Pan, E.; Bogumil, D.; Cortessis, V.; Yu, S.; Nieva, J. A Systematic Review of the Efficacy of Preclinical Models of Lung Cancer Drugs. Front. Oncol. 2020, 10, 591. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cancer Type | Source of Organoids | Culture Technique | Endpoint of Study | Resemblance to Parent Tumor | References |
|---|---|---|---|---|---|
| Bladder cancer | Human bladder cancer cells | Matrigel + culture with hepatocyte medium, charcoal-stripped serum, and ROCK inhibitor | Patient-derived bladder cancer organoids able to recapitulate histologic findings of primary tumors | Histopathologic analysis of tumor organoids showed concordance to parental tumors. | Lee et al. (2018) [59] |
| Breast cancer | Human breast cancer single cells | Culture with medium including mitogen Neuregulin 1 | Protocol for breast cancer organoid creation | Blinded histologic analysis showed concordant lobular and ductal carcinoma and breast cancer biomarkers. | Sachs et al. (2018) [28] |
| Human breast cancer tissue | Matrigel or 3D collagen I + culture with medium containing insulin, EGF, hydrocortisone, and cholera toxin | Identified expression of cytokeratin-14 and p63 important for invasion of poorly differentiated carcinomas | Cytokeratin-14+ cells were observed in primary tissue and corresponding organoids. | Cheung et al. (2013) [73] | |
| Normal and tumor breast cancer cells | Three-dimensional bioprinting | Three-dimensional bioprinting able to create multicellular, architecturally defined, scaffold-free organoid models | Bioprinted organoids retained a similar structural morphology to the parental tumor. | Langer et al. (2019) [74] | |
| Clear-cell renal cell carcinoma | Human clear-cell renal cell carcinoma single cells | Kidney three-dimensional medium + Matrigel and cultured for organoid formation | Patient-derived clear-cell renal cell carcinoma organoids demonstrating a high degree of genetic similarity to primary tissue | PDOs were concordant to primary tissue through VHL sequencing. | Bolck et al. (2019) [75] |
| Gastrointestinal tract cancers | Mouse- and human-derived adult stem cells, adult epithelial crypts, and colon cancer cells | Matrigel + culture media containing human Wnt-3A | Refined protocol for the culture and long-term culture of colon adenocarcinoma organoids | Similar levels of LGR5 and Axin2 were observed on day 7 of organoid culture compared to fresh adenoma tissue. | Sato et al. (2009) [36] Sato et al. (2011) [48] |
| Mouse and human adult stem cells and mouse neonatal gut | Organoids derived from genetically engineered mice using a single air–liquid interface culture approach | Gastrointestinal primary tissue epithelial and mesenchymal organoids reprogrammed to model cancer formation and oncogene activation | Organoids displayed a well-differentiated epithelial layer of surface mucous cells. | Li et al. (2014) [76] | |
| Human induced pluripotent stem cells | CRISPR/Cas9-mediated genome editing to normal human small-intestinal organoid stem cell cultures | Creation of colorectal cancer organoids through genetic modification of normal intestinal epithelium organoids to study disease | Orthotopically implanted genetically engineered organoids displayed large cysts and well-differentiated carcinomas. | Drost et al. (2015) [49] Matano et al. (2014) [50] | |
| Human colorectal cancer tissue | Followed Sato et al.’s (2011) protocol | Tested the sensitivity of cetuximab based on molecular characterization of patient-derived colorectal cancer organoids | There was no direct comparison to primary tissue. | Verissimo et al. (2016) [51] | |
| Human colorectal cancer tissue | Followed Sato et al.’s (2011) protocol | Patient-derived colorectal tumor organoids protocol able to recapitulate somatic copy number and mutations found in colorectal cancer primary tissue | All studies found concordance of genetic diversity in organoids and parent tumor with DNA sequencing. | Van de Wetering et al. (2015) [30] Weeber et al. (2015) [52] Roerink et al. (2018) [46] | |
| Human esophageal adenocarcinoma cells | BME + culture | Patient-derived esophageal adenocarcinoma organoid protocol created | Organoids displayed normal glandular architecture and wild-type p53 expression consistent with parent gastric tissue. | Li et al. (2018) [55] | |
| Human gastric cancer cells | Matrigel + culture with media supplemented with ROCK inhibitor Y-27632 | Patient-derived gastric cancer organoids having similar immunohistochemical and mutational profiles as primary gastric cancer tissue | PDOs displayed cytokeratin 7, cadherin 17, carcinoembryonic antigen, and periodic acid Schiff reaction similar to parent tissue. | Seidlitz et al. (2017) [77] | |
| Human gastric cancer tissue | Established based on Barfeld et al.’s (2015) protocol; cultured in media containing Nutlin-3, ROCK inhibitory free medium, TGF-β, and absence of EGF and FGF-10 | Patient-derived gastric cancer organoid biobank established to investigate the role of driver gene mutations in gastric cancer | Histologic and genomic analysis of the organoid displayed concordance to parent tissue. | Nanki et al. (2018) [78] | |
| CDH1 and Trp53 murine gastric cancer tissue | Collagen gel + double-dish air–liquid interface culture system | Murine-derived gastric cancer organoids to study the role of TGF-β receptor 2 loss of function mutation in metastatic transformation of diffuse gastric cancer | Organoids displayed similar copy number variation, allelic imbalances, and rearrangements as primary tissue. | Nadauld et al. (2014) [64] | |
| Human gastrointestinal cancer cells | Matrigel + culture with human organoid media | Established organoid biobank for patient-derived metastatic and/or relapsed colorectal and gastroesophageal cancer | Colorectal organoids retained diffuse and intestinal growth patterns. | Vlachogiannis et al. (2018) [79] | |
| Liver cancer | Human primary liver cancer cells | BME + culture with media containing 3 nM dexamethasone | Hepatocellular carcinoma, cholangiocarcinoma, and combined hepatocellular carcinoma/cholangiocarcinoma organoid model creation | Organoids formed pseudo-glandular rosettes consistent with parent hepatocellular carcinoma. | Broutier et al. (2017) [25] |
| Non-small-cell lung cancer | Human non-small-cell lung cancer cells | BME + culture with media containing Nutlin-3a used to select for specific cancer organoids | Demonstrated the ability of lung tumor organoids to recapitulate histopathologic and genetic features of primary tissue | Organoids recapitulated histopathologic features of adenocarcinoma, mucinous adenocarcinoma, and large-cell neuroendocrine parent tumors. | Sachs et al. (2019) [65] |
| Ovarian cancer | Human induced pluripotent stem cells | Matrigel + cultured according to protocols by Xia et al., May et al., and Takasoto et al. | Conditions to create fallopian tube epithelial organoids described to study high-grade serous ovarian carcinoma | Organoids expressed TUB4A, FOXJ1, and PAX8 consistent with normal fallopian tubes. | Yucer et al. (2017) [47] |
| Human ovarian cancer cells | BME + culture in media containing hydrocortisone, forskolin, and heregulin β-1 | Protocol established for creation and propagation of ovarian cancer organoids | Expression of markers of secretory and ciliated cells was shared with primary tissue. | Kopper et al. (2019) [27] | |
| Pancreatic cancer | Murine pancreatic ductal cells and human malignant pancreatic tissue | Matrigel + culture with media containing Wnt3a, Noggin, epidermal growth factor, gastrin, fibroblast growth factor 10, gastrin, nicotinamide, and A83-01 | Culture conditions refined to passage and transplant pancreatic organoids for molecular and cellular biology analysis | PDOs had elevated CA 19-9 expression consistent with parent tumor tissue. | Boj et al. (2015) [24] |
| Human pancreatic ductal cancer tissue | Culture with pancreatic progenitor and tumor organoid medium | Conditions refinedto support tumor organoid growth from patient-derived pancreatic ductal adenocarcinoma organoids | Organoids expressed NKX6.1 and PTF1A, pancreas-specific markers, on gene expression analysis. | Huang et al. (2015) [80] | |
| Human pancreatic adenocarcinoma cells | Matrigel + culture with media containing Nutlin-3 and BMP-4, absent of EGF and Noggin | Pattern of dependency on Wnt ligands determined for pancreatic cancers using patient-derived organoids | Organoids contained common driver gene alterations, including KRAS, CDKN2A, TP53, and SMAD4, on whole-exome sequencing and comparative genomic hybridization analyses. | Seino et. Al. (2018) [81] | |
| Human pancreatic cancer cells | Matrigel + culture in human complete feeding medium | Establishment of a patient-derived pancreatic cancer organoid library | Whole-exome sequencing of organoids and PDOs was performed. | Tiriac et al. (2018) [66] | |
| Prostate cancer | Genetically engineered murine prostate epithelial cells and human prostatic epithelium | Matrigel + culture with media containing EGF, Noggin, R-spondin 1, FGF-10, FGF-2, prostaglandin E2, nicotinamide, and p38 inhibitor SB202190 | Creation of a culture system for long-term expansion of murine and human prostatic epithelial organoids | Organoids displayed a cystic structure and expressed basal prostate markers, including p63, Ck5, and Ck8. | Karthaus et al. (2014) [60] |
| Human induced pluripotent stem cells | Matrigel + culture with growth factors | Prostate cancer organoid protocol description | PDOs maintained typical histologic patterns of prostate adenocarcinomas. | Gao et al. (2014) [26] |
| Cancer Type | Source of Organoids | Culture Technique | Endpoint of Study | Resemblance to Parent Tumor | References |
|---|---|---|---|---|---|
| Glioblastoma | Patient-derived glioma stem cells | 1. Co-culture of GSCs and iPSCs 2. Supplementing GSCs with normal cerebral organoids 3. Fusion of GSC spheres with normal brain organoids | Three techniques to model GSC invasion in normal brain organoids and creation of GBM organoids | No direct comparison to primary tissue could be made. | Goranci-Buzhala et al. (2020) [83] |
| Patient-derived glioblastoma and non-glioblastoma stem cells | Matrigel + culture shaking in NBM complete media | Spatial distribution of GBM replicated in organoids derived from glioblastoma and non-glioblastoma stem cells | Orthotopically implanted PDOs were diffuse and infiltrative, histologically resembling the parent tumor. | Hubert et al. (2016) [57] | |
| Human embryonic stem cells | Normal cerebral organoids co-cultured with patient-derived tumor cells or oncogene introduction through electroporation | Normal human-derived cerebral organoids a vector for glioblastoma organoid modeling | Engineered PDOs displayed a mesenchymal phenotype consistent with patient-derived GBMs on transcriptomic analysis. | Ogawa et al. (2018) [56] | |
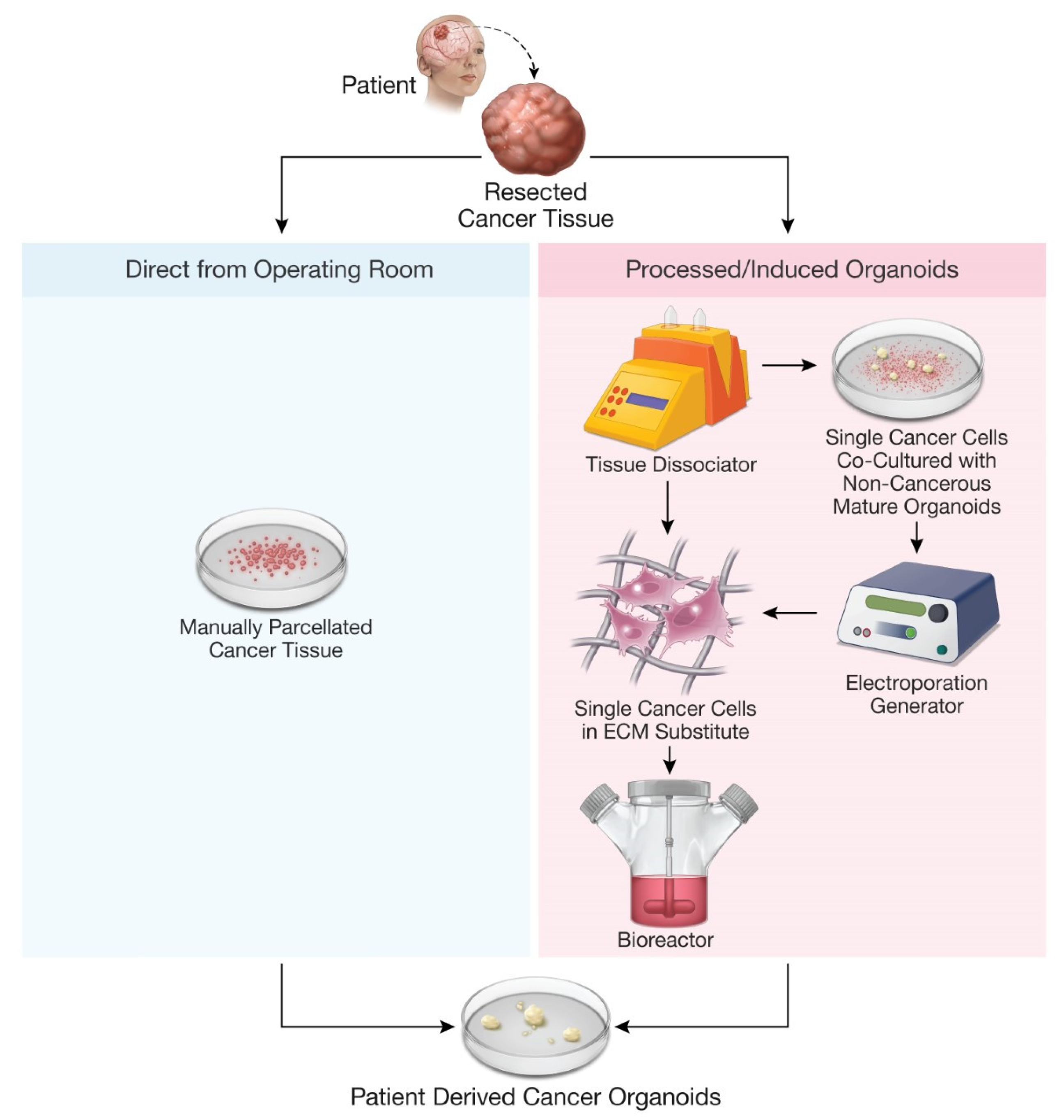
| Human glioblastoma tissue | Patient tissue parcellated and cultured without an extracellular matrix | Glioblastoma organoid protocol development from primary tissue samples with minimum processing | PDOs had cellular and nuclear atypia, abundant mitotic figures, and pleomorphic nuclei consistent with high-grade parent tumors on histologic analysis. | Jacob et al. (2020) [62] | |
| Glioblastoma spheroids infiltrating cerebral organoids | Co-culture of mouse early (6-day-old) cerebral organoids with glioblastoma spheroids created from glioblastoma stem cell culture | Demonstration of hybrid glioblastoma organoid modeling | Co-cultured organoids displayed core infiltration and expression of GBM stem cell markers NESTIN and SOX2. | Da Silva et al. (2018) [82] | |
| Patient-derived glioma stem cells | Co-culture of cerebral organoids with transduced GSCs on NBM | Single-cell RNA sequencing comparison of four patient-derived glioblastoma models | Organoids displayed microscopic invasion and single-cell heterogeneity consistent with parent tumors. | Pine et al. (2020) [85] | |
| Central nervous system primitive neuroectodermal-like and glioblastoma-like tumor | Human embryonic stem cells | Neoplastic cerebral organoids (Neo-COR); combination of plasmids introduced into normal cerebral organoids through electroporation before te organoids embedded in Matrigel | Demonstration of brain tumorigenesis through introduction of oncogenic mutations in normal cerebral organoids through transposon and CRISPR/Cas9 mutagenesis | GBM-like Neo-COR displayed upregulation of GBM genes on transcriptomic analysis. | Bian et al. (2018) [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pernik, M.N.; Bird, C.E.; Traylor, J.I.; Shi, D.D.; Richardson, T.E.; McBrayer, S.K.; Abdullah, K.G. Patient-Derived Cancer Organoids for Precision Oncology Treatment. J. Pers. Med. 2021, 11, 423. https://doi.org/10.3390/jpm11050423
Pernik MN, Bird CE, Traylor JI, Shi DD, Richardson TE, McBrayer SK, Abdullah KG. Patient-Derived Cancer Organoids for Precision Oncology Treatment. Journal of Personalized Medicine. 2021; 11(5):423. https://doi.org/10.3390/jpm11050423
Chicago/Turabian StylePernik, Mark N., Cylaina E. Bird, Jeffrey I. Traylor, Diana D. Shi, Timothy E. Richardson, Samuel K. McBrayer, and Kalil G. Abdullah. 2021. "Patient-Derived Cancer Organoids for Precision Oncology Treatment" Journal of Personalized Medicine 11, no. 5: 423. https://doi.org/10.3390/jpm11050423
APA StylePernik, M. N., Bird, C. E., Traylor, J. I., Shi, D. D., Richardson, T. E., McBrayer, S. K., & Abdullah, K. G. (2021). Patient-Derived Cancer Organoids for Precision Oncology Treatment. Journal of Personalized Medicine, 11(5), 423. https://doi.org/10.3390/jpm11050423