3. Methods
The CancerNext® +RNAinsight® germline test at Ambry Genetics lab provided an analysis of 34 genes associated with hereditary cancer predisposition. Genomic deoxyribonucleic acid (gDNA) and ribonucleic acid (RNA) were isolated from the patient specimen using standardized methodology and quantified. RNA was converted to complementary DNA (cDNA) by reverse-transcriptase polymerase chain reaction (RT-PCR). Sequence enrichment of the targeted coding exons and adjacent intronic nucleotides was carried out via a bait-capture methodology using long biotinylated oligonucleotide probes followed by polymerase chain reaction (PCR) and next-generation sequencing. Additional DNA analyses included Sanger sequencing for any regions missing or with insufficient read depth coverage for reliable heterozygous variant detection. Variants in regions complicated by pseudogene interference, variant calls not satisfying depth of coverage and variant allele frequency quality thresholds, and potentially homozygous variants were verified by Sanger sequencing. The BRCA2 Portuguese founder mutation, c.156_157insAlu (also known as 384insAlu), and the MSH2 coding exons 1–7 inversion were detected by next-generation sequencing and confirmed by multiplex ligation-dependent probe amplification (MLPA) or PCR and agarose gel electrophoresis. Gross deletion/duplication analysis for 30 of the genes (excluding HOXB13, PMS2, POLD1, and POLE) was performed using a custom pipeline based on read depth from NGS data and/or targeted chromosomal microarray with confirmatory MLPA when applicable. Gross deletion/duplication analysis of PMS2 was performed using MLPA kit P008-B1. If a deletion was detected in exons 13, 14, or 15 of PMS2, double-stranded sequencing of the appropriate exon(s) of the pseudogene PMS2CL was performed to determine whether the deletion was located in the PMS2 gene or pseudogene. All sequence analysis was based on the following NCBI reference sequences: APC- NM_000038.5 & NM_001127511.2, ATM- NM_000051.3, BARD1- NM_000465.2, BMPR1A- NM_004329.2, BRCA1- NM_007294.3, BRCA2- NM_000059.3, BRIP1- NM_032043.2, CDH1- NM_004360.3, CDK4- NM_000075.3, CDKN2A- NM_000077.4 and NM_058195.3 (p14ARF), CHEK2- NM_007194.3, DICER1- NM_177438.2, HOXB13- NM_006361.5, MUTYH- NM_001128425.1, MRE11A NM_005591.3, MLH1- NM_000249.3, MSH2- NM_000251.1, MSH6- NM_000179.2, NBN- NM_002485.4, NF1- NM_000267.3, PALB2- NM_024675.3, PMS2- NM_000535.5, POLD1- NM_002691.2, POLE- NM_006231.2, PTEN- NM_000314.4, RAD50- NM_005732.3, RAD51C NM_058216.1, RAD51D- NM_002878.3, SMAD4- NM_005359.5, SMARCA4- NM_001128849.1, STK11- NM_000455.4, TP53- NM_000546.4.
Analytical range: The CancerNext® +RNAinsight® test was used to target detection of DNA sequence mutations in 32 genes (APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, DICER1, HOXB13, MLH1, MSH2, MSH6, MUTYH, MRE11A, NBN, NF1, PALB2, POLD1, POLE, PMS2, PTEN, RAD50, RAD51C, RAD51D, SMAD4, SMARCA4, STK11, and TP53) by either next-generation or Sanger sequencing of all coding domains and well into the flanking 5′ and 3′ ends of all the introns and untranslated regions. For HOXB13, only variants impacting codon 84 are routinely reported. For POLD1 and POLE, only missense and in-frame indel variants in the exonuclease domains (codons 311–541 and 269–485, respectively) are routinely reported. Gross deletion/duplication analysis determines gene copy number for the covered exons and untranslated regions of sequenced genes (excluding HOXB13, POLD1, and POLE) as well as GREM1 and EPCAM. For GREM1, only the status of the 40 kb 5′UTR gross duplication is analyzed and reported. For EPCAM, only gross deletions encompassing the 3′ end of the gene are reported. For APC, all promoter 1B gross deletions as well as single-nucleotide substitutions within the promoter 1B YY1 binding motif (NM_001127511 c.-196_-186) are analyzed and reported. RNA transcripts were screened for 18 genes (APC, ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NF1, PALB2, PMS2 exons 1–10, PTEN, RAD51C, RAD51D, and TP53) and compared to a human reference pool. The absence or presence of RNA transcripts meeting quality thresholds were incorporated as evidence towards assessment and classification of DNA variants. Any regions not meeting RNA quality thresholds were excluded from analysis. Regions routinely excluded due to chronically low expression in human peripheral lymphocytes include: BRCA2 (exon 1), BRIP1 (exons 18, 20), CDH1 (exons 1, 2, 16), and CHEK2 (exons 1, 7, 8).
Tempus Lab PD-L1 tumor staining was performed on the endometrial and ovarian tissue samples. PD-L1 is defined as complete circumferential and/or partial linear plasma membrane staining of tumor cells at any intensity. Tumor-associated immune cell staining was defined as membrane and/or cytoplasmic staining (at any intensity) of mononuclear inflammatory cells (MICs) within tumor nests and adjacent supporting stroma. PD-L1 IHC 22C3 pharmDx is a qualitative immunohistochemical assay using Monoclonal Mouse Anti-PD-L1, Clone 22C3 intended for use in the detection of PD-L1 protein in formalin-fixed, paraffin-embedded (FFPE) non-small-cell lung cancer (NSCLC), gastric or gastroesophageal junction (GEJ) adenocarcinoma, esophageal squamous cell carcinoma, cervical cancer, and urothelial carcinoma tissues. Scoring is not provided in tumors for which no scoring system has been published. See the KEYTRUDA® product label for expression cutoff values guiding therapy in specific clinical circumstances. The same DAKO PD-L1 22C3 clone was used for both tumors.
The initial IHC for MMR protein expression test was run at TriHealth pathology lab on an endometrial sample from a total abdominal hysterectomy with bilateral salpingo-oophorectomy. This demonstrated intact nuclear expression for MLH1, MSH2, MSH6, and PMS2. Background nonneoplastic tissue/internal control was run with intact nuclear expression. A second IHC for MMR protein expression was run at Tempus Labs using the same endometrial tissue block.
Somatic tumor profiling was performed by Tempus Labs on endometrial and ovarian tissue samples, with an accompanying matched normal blood sample. The Tempus|xT next-generation sequencing assay is a CAP/CLIA-validated panel designed to detect actionable oncological targets by sequencing FFPE tumor samples with matched normal saliva or blood samples, when available. The Tempus|xT assay includes DNA sequencing of 648 genes spanning ~3.6 Mb of genomic space and full-transcriptome RNA sequencing. From DNA sequencing, somatic and incidentally detected germline single-nucleotide variants (SNVs), insertions and deletions (indels), and copy number variants (CNVs) are detected. Additionally, translocations in 22 genes are detected, along with two promoter regions (
PMS2 and
TERT) and 239 sites used to determine microsatellite instability (MSI) status. Tumor mutational burden (TMB) is calculated as described below. Some viral sequences, such as HPV and EBV, may be reported to offer a diagnostic or prognostic insight when deemed appropriate by Tempus pathologists. From RNA-seq, gene fusions (translocations) are detected in an unbiased and comprehensive manner. Full-transcriptome RNA expression counts are analytically validated. The Tempus|xT assay requires specimens with a tumor content of 20% post macrodissection (minimum 30% for MSI status). Clinical sequencing is performed to 500× depth of coverage for tumor specimens and 150× for normal specimens. Performance specifications and a complete gene list are available online at
https://www.tempus.com/genomic-profiling/#proprietary-sequencing (accessed on 15 April 2021).
The Tempus|xT assay identifies variants by aligning the patient’s DNA sequence to the human genome reference sequence version hg19 (GRCh37) and classifies each variant as potentially actionable, biologically relevant, variant of unknown significance (VUS), or benign. Variants considered potentially actionable alterations are protein-altering variants with an associated therapy based on evidence from the medical literature. Biologically relevant alterations are protein-altering variants that may have functional significance or have been observed in the medical literature, but are not associated with a specific therapy in the Tempus knowledge database. VUSs are protein-altering variants exhibiting an unclear effect on function and/or without sufficient evidence to determine their pathogenicity. Benign variants are not reported. The clinical summary shows actionable and biologically relevant somatic variants, and certain pathogenic or likely pathogenic inherited variants that are reported as incidental findings when applicable. Reportable secondary/incidental findings are limited to genes and variants associated with inherited cancer syndromes. Germline genes that are reported include: APC, ATM, AXIN2, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDKN2A, CEBPA, CHEK2, EGFR, EPCAM, ETV6, FH, FLCN, GATA2, MEN1, MLH1, MSH2, MSH3, MSH6, MUTYH, NBN, NF2, PALB2, PMS2, POLD1, POLE, PTEN, RAD51C, RAD51D, RB1, RET, RUNX1, SDHAF2, SDHB, SDHC, SDHD, SMAD4, STK11, TP53, TSC1, TSC2, VHL, and WT1.
TMB calculated by the Tempus|xT assay measures the quantity of somatic mutations of any pathogenicity, including benign, carried in a tumor as the number of single-nucleotide protein-altering mutations per million coding base pairs. TMB is calculated at the time of initial report delivery. Accordingly, the TMB calculation is based upon (a) both the tumor and normal sample if Tempus had analyzed both at the time of the initial report, or (b) the tumor sample only if no normal sample had been analyzed at the time of the initial report. MSI refers to hypermutability caused by genetic or acquired defects in the DNA mismatch repair pathway. MSI status is divided into MSI-high (MSI-H), microsatellite stable (MSS), and microsatellite equivocal (MSE) tumors. MSI-H tumors have changes in microsatellite repeat lengths due to defective DNA mismatch repair activity, MSS tumors do not have detectable defects in DNA mismatch repair, and MSE tumors have an intermediate phenotype which cannot be clearly classified as MSI-H or MSS based on the statistical cutoff used to define those categories. If MSI status will affect clinical management, immunohistochemical staining for DNA mismatch repair proteins, or application of another method for ascertaining MSI status, is recommended.
Homologous recombination deficiency (HRD) status was determined for the endometrial and ovarian tissue samples using the Tempus|HRD assay. The Tempus|HRD assay computational algorithm uses results from tumor and normal matched xT sequencing data to calculate the genome-wide loss of heterozygosity (GWLOH) percentage and uses the somatic and germline alteration status of BRCA1 and BRCA2 to determine HRD status. GWLOH is calculated by determining the percentage of genomic segments with LOH by the Tempus copy number calling algorithm (CONA). GWLOH is considered positive for HRD at ≥29% for breast cancer, ≥25% for ovarian cancer, ≥28% for pancreatic cancer, and ≥33% for any other cancer type (endometrial). BRCA1 and BRCA2 alterations considered positive for HRD include the following: a pathogenic or likely pathogenic alteration with LOH, biallelic pathogenic or likely pathogenic alterations, or two-copy loss. BRCA1/2 LOH is computed using CONA, which uses tumor purity and copy states in the tumor genome to generate copy number status. HR-pathway genes analyzed on the Tempus|xT panel include: ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FANCL, HDAC2, MRE11, NBN, PALB2, RAD51B, RAD51C, RAD51D, and RAD54L.
4. Results
Family history included three cases of breast cancer in early 50s, pancreatic, and a gastrointestinal cancer. There were no known cases of colon cancer or other gynecological cancers (
Figure 1). Patient met with a genetic counselor (RS) and consented to germline genetic testing via a 34 gene germline cancer panel at Ambry Genetics lab. Testing identified a germline likely pathogenic
PMS2 mutation c.2095G>C p.D699H mutation and a
PALB2 VUS c.1250C>A p.S417Y.
Patient underwent germline testing at Ambry Genetics lab with a 34 gene CancerNext +RNAinsight
® panel, identifying a likely pathogenic
PMS2 mutation and a
PALB2 VUS. This
PMS2 mutation is classified as likely pathogenic on ClinVar. The location of the
PMS2 germline mutation c.2095G>C p.D699H mutation was at the 3′ end of the
PMS2 gene in exon 12. The
PMS2 mutation was classified as likely pathogenic in ClinVar database. This is an area with high homology to the
PMS2CL pseudogene [
14]. The Tempus next-generation sequencing (NGS) assay is unable to distinguish with certainty
PMS2 from
PMS2CL, so the
PMS2 pathogenic result was not reported on the limited set of germline genes reported by Tempus. To distinguish the pseudogene, Ambry Genetics used a combination of long-range PCR and a multiplex ligation-dependent PCR amplification (MLPA). The isoforms used by each laboratory are available by request.
The germline
PALB2 variant was classified as a VUS based on the following information from Ambry lab: The p.S417Y variant (also known as c.1250C>A), located in coding exon 4 of the
PALB2 gene, results from a C to A substitution at nucleotide position 1250. The serine at codon 417 is replaced by tyrosine, an amino acid with dissimilar properties. Functional studies have demonstrated that S417Y leads to a partial reduction of ChAM-mediated PALB2 chromatin association without affecting the cellular resistance to CPT [
15]. This amino acid position is highly conserved in available vertebrate species. In addition, the in silico prediction for this alteration was inconclusive. Since supporting evidence is limited at this time, the clinical significance of this alteration remains unclear.
Because the proficient MMR result on endometrial IHC for MMR expression was not consistent with the germline PMS2 Lynch mutation, the clinical team (KH, DD) offered somatic tumor profiling to better understand this patient’s tumors and to determine whether this tumor was indeed Lynch-associated. The patient consented to Tempus|xT solid tumor profiling with a matched tumor normal blood sample. The tumor sample from the left fallopian tube and ovary of mixed endometrioid adenocarcinoma and clear-cell carcinoma and the uterine specimen of endometrioid adenocarcinoma were both sent to Tempus for xT tumor profiling. Tempus|xT targeted somatic tumor profiling of 648 genes was performed. This included IHC for PD-L1 expression (
Figure 2) and MMR (
Figure 3), TMB, homologous recombination deficiency (HRD), somatic, and germline analysis.
TMB was 245.8 mut/MB in ovarian and 330.5 mut/MB in endometrial with high microsatellite instability status (MSI-H) for both tumors. There was no recognized second hit, or mutation, identified in
PMS2. This very high TMB is considered an ultrahypermutator tumor by Campbell et al. [
16]. It was noted that a
POLE variant was identified on tumor profile testing in both tumors (
POLE c.1306C>T p.P436S) and that polymerase proofreading alterations in
POLE can play a role in ultrahypermutator phenotypes. The
POLE c.1306C>T p.P436S variant was reviewed by a Tempus Lab variant scientist and functional evidence led to a somatic pathogenic classification. The evidence used to classify
POLE c.1306C>T p.P436S as pathogenic included:
1. Variant causes increased mutagenesis in yeast assays [
17]; variant is referred to as p.P451S in yeast).
2. Variant identified in a patient whose phenotype is similar to patients with known
POLE mutations [
18].
3. Variant falls in the
POLE exonuclease domain (codons 269–485). While this evidence is sufficient to classify the variant as pathogenic in a somatic context, it does not quite meet the threshold for being classified pathogenic as a germline variant and is seen as germline VUS in ClinVar. We cannot be certain that the ultrahypermutator phenotype was caused by
PMS2,
POLE, or a synergistic effect. The fact that the patient had such a high TMB could be consistent with this
POLE variant driving the hypermutator phenotype. Additionally, the
POLE mutation is associated with high proportions of C>A, C>T, and T>G variants [
19], and a large number of these were observed in this patient’s case. Alexandov has described mutational signatures caused by mutations in the exonuclease domain of
POLE, including single-base substitutions SBS10a, SBS10b, and SBS28, referenced in the
Catalogue of Somatic Mutations in Cancer. Tumors with these mutational profiles generate a large volume of somatic mutations (>100 mut/MB) and are termed hypermutators [
20,
21,
22].
MLH1 promoter hypermethylation is not a standard component of Tempus xT testing.
BRAFV600E was included in the somatic tumor profiling and was not identified. IHC assessment of MMR gene protein expression was also performed and was reported as normal on both tumors. The immunohistochemical staining performed at Tempus Labs noted that
PMS2 expression was weak in the uterine tumor, but was still considered normal (
Figure 3).
Both tumors had a very large number of somatic variants. The ovarian tumor had 5 potentially actionable and 21 biologically relevant variants. The uterine tumor had 7 potentially actionable and 27 biologically relevant variants (
Figure 4). In addition, the ovarian tumor had 506 VUSs and the endometrial tumor had 657 VUSs. When variants were compared between tumors, there were many variants shared between them. For example, there were three potentially actionable somatic variants in common between the ovarian and uterine tumors:
BRCA1 p.W312*,
ATM p.I1270fs, and
PIK3CA p.R93Q. The
POLE p.P436S variant was also seen in both tumors but was classified as potentially actionable in the uterine cancer and as a VUS in the ovarian cancer. There were also 13 shared variants among the biologically relevant variants (
Figure 4). The ovarian tumor had 26 potentially actionable and biologically relevant variants, of which 16 (61.5%) were in common with the endometrial tumor. The endometrial tumor had 34 potentially actionable and biologically relevant variants, of which 16 (47%) were in common with the ovarian tumor. It was confirmed by Ambry Genetics that none of these other common gene variants, among the 34 genes tested at Ambry, were germline.
Homologous recombination deficiency (HRD) was also evaluated via genome-wide loss of heterozygosity (LOH) in each tumor (
Figure 5). While both tumors demonstrated pathogenic/likely pathogenic genomic variants in the HRD pathway (
BRCA1/2 and
ATM), the genome-wide loss of heterozygosity (LOH) was well below the threshold of 33% and was deemed “not detected”.
The tumor profile report gave multiple therapy options for this patient, including pembrolizumab, which is FDA-approved for the current diagnosis based on MSI-H and TMB, PARP inhibitor based upon somatic BRCA1, BRCA2, and ATM variants, and alpelisib based on the PIK3CA variant. In addition, the uterine tumor report had FDA-approved therapies for other indications: everolimus and sirolimus for mTOR inhibitors and nivolumab based on the POLE variant.
5. Discussion
This patient had a personal and family history of cancer that was not consistent with the moderate-risk
PMS2 Lynch syndrome risk profile in NCCN [
3]. The Prospective Lynch Syndrome Database has reported primarily colorectal and endometrial cancer risk to be associated with
PMS2 [
23]. This patient’s personal and family history included ovarian cancer, three cases of breast cancer, and pancreatic cancer. These are all at lower risk in
PMS2 germline carriers, but breast cancers have been reported in some
PMS2 Lynch patients [
24]. Although
PMS2 mutation carriers do have lower overall cancer risk compared to other Lynch mutations carriers, a smaller study of
PMS2 LS patients showed that 60% of patients with MMR-deficient/MSI tumors presented with extracolonic cancers [
25]. The NCCN guidelines primarily address surveillance and prevention strategies for colon and gynecological cancers with
PMS2 germline mutation. Clinicians may consider family history to determine any indication for screening other cancers.
The location of the PMS2 mutation c.2095G>C p.D699H mutation was in an area with high homology to the PMS2CL pseudogene. The tumor profiling lab was unable to distinguish the germline PMS2 from pseudogene, demonstrating the importance of high-quality germline analysis beyond sequencing. In this case, both the germline and the somatic lab provided crucial information. Ambry Genetics lab reported that in vitro studies of this 3′ mutation showed expression levels similar to wildtype with reduced protein function. This likely explains why PMS2 protein expression was proficient on IHC in this patient’s tumor, albeit weakly. Despite the PMS2 expression on IHC, the tumors both had high TMB and MSI-H, confirming that these tumors were both Lynch-associated.
The high TMB in both tumors was categorized as ultrahypermutator [
16]. It was noted that the TMB in the endometrial tumor was higher at 330.5 mut/MB than the ovarian at 245.8 mut/MB, and that a
POLE variant was present in both tumors. This very high TMB may have been caused by the germline
PMS2 mutation, and the somatic
POLE polymerase proofreading alteration may also have played a role in tumorigenesis.
The ability to compare somatic mutations shared by the endometrial and the ovarian tumors gave insight to a shared origin. The ovarian tumor had 16/26 or 61.5% potentially actionable and biologically relevant variants in common with the endometrial tumor, and the endometrial tumor had 16/34 or 47% of potentially actionable and biologically relevant variants in common with the ovarian tumor. Both tumors shared three potentially actionable variants in
ATM,
BRCA1 and
PIK3CA, as well as 13 biologically relevant variants in
APC,
ATR,
CREBBP,
CTCF,
HNF1A,
KEAP1,
KMT2D,
NCOR1,
NF1,
PTEN,
PTPN13,
TBX3, and
TP53 (
Figure 4). In addition to the potentially actionable and biologically relevant variants, the number of VUSs was also reported for each tumor. The ovarian tumor had 506 VUSs and the endometrial tumor had 657 VUSs. We were unable to compare the VUSs directly for commonality.
Deshpande et al. demonstrated that better understanding of the pathways leading to MSI-high gynecological cancers will improve prediction of cancer progression and therapeutic response [
26]. Takeda et al. stated that most clinically diagnosed cases of SEOC have clonally related cancers, indicating metastatic cancer [
12]. Niskakoski et al. performed deep sequencing of 578 genes in five synchronous Lynch carcinomas with germline
MLH1 and
MSH2 [
27]. The group found that synchronous cancers were concordant molecularly, suggesting shared origins in SEOC in Lynch syndrome. Moukarzel evaluated a series of five patients with germline MMR, looking at the clonal relationship and directionality of progression in SEOC and comparing to patients with sporadic SEOC [
13]. They concluded that the directionality of progression was likely from the endometrium to the ovary. They found evidence that SEOC in LS patients may represent distinct primary tumors, which is logical considering the genetic predisposition. However, Moukarzel et al. did conclude that a subset of LS-associated SEOCs may originate from a single primary tumor, with endometrium being the most likely origin. Our case is a rare example of SEOC with germline
PMS2 mutation with 16 shared actionable and biologically relevant somatic variants that demonstrate clonality of the two carcinomas arising from one common site, most likely the endometrium.



 and
and 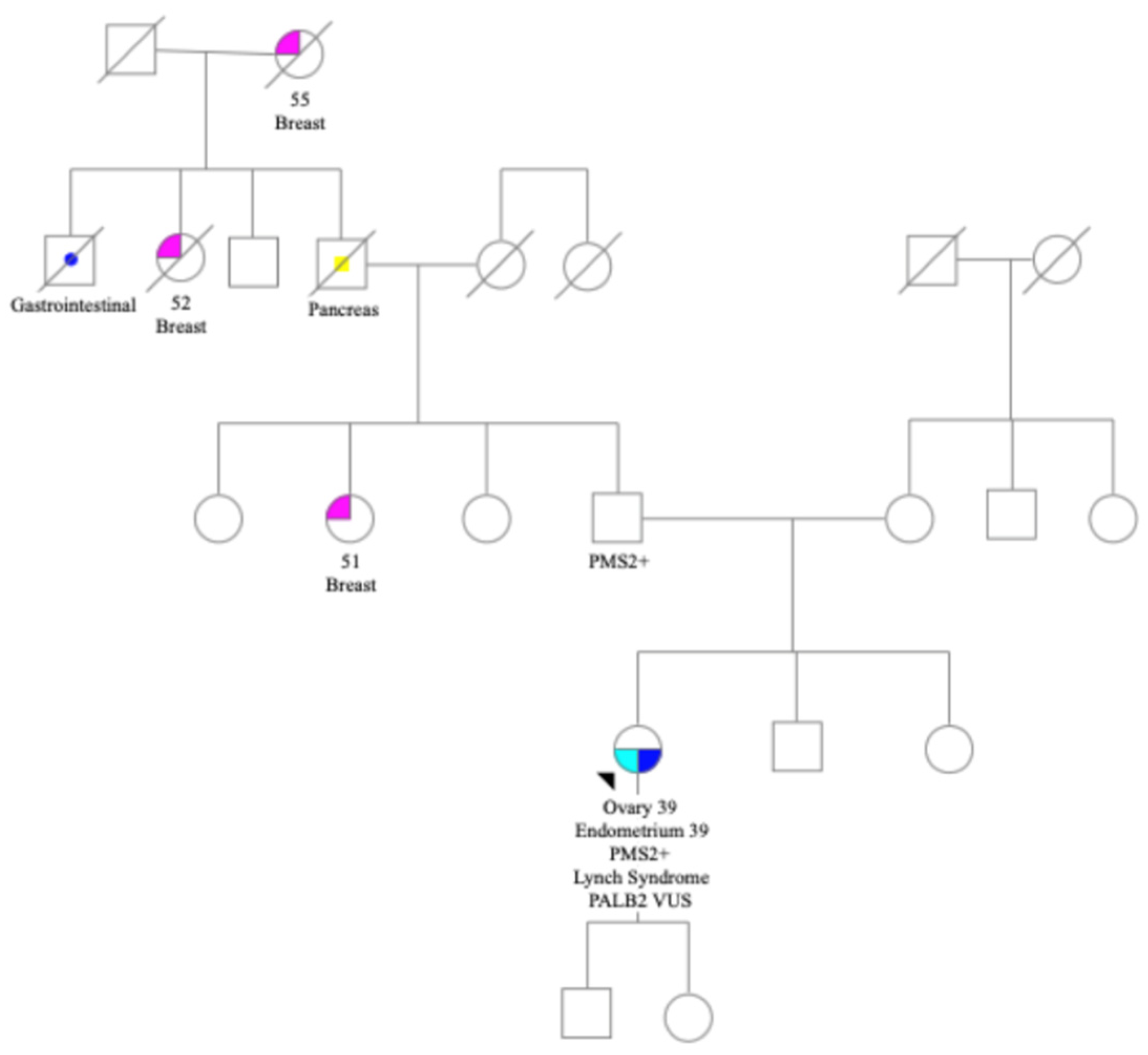{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




