
Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia



Abstract
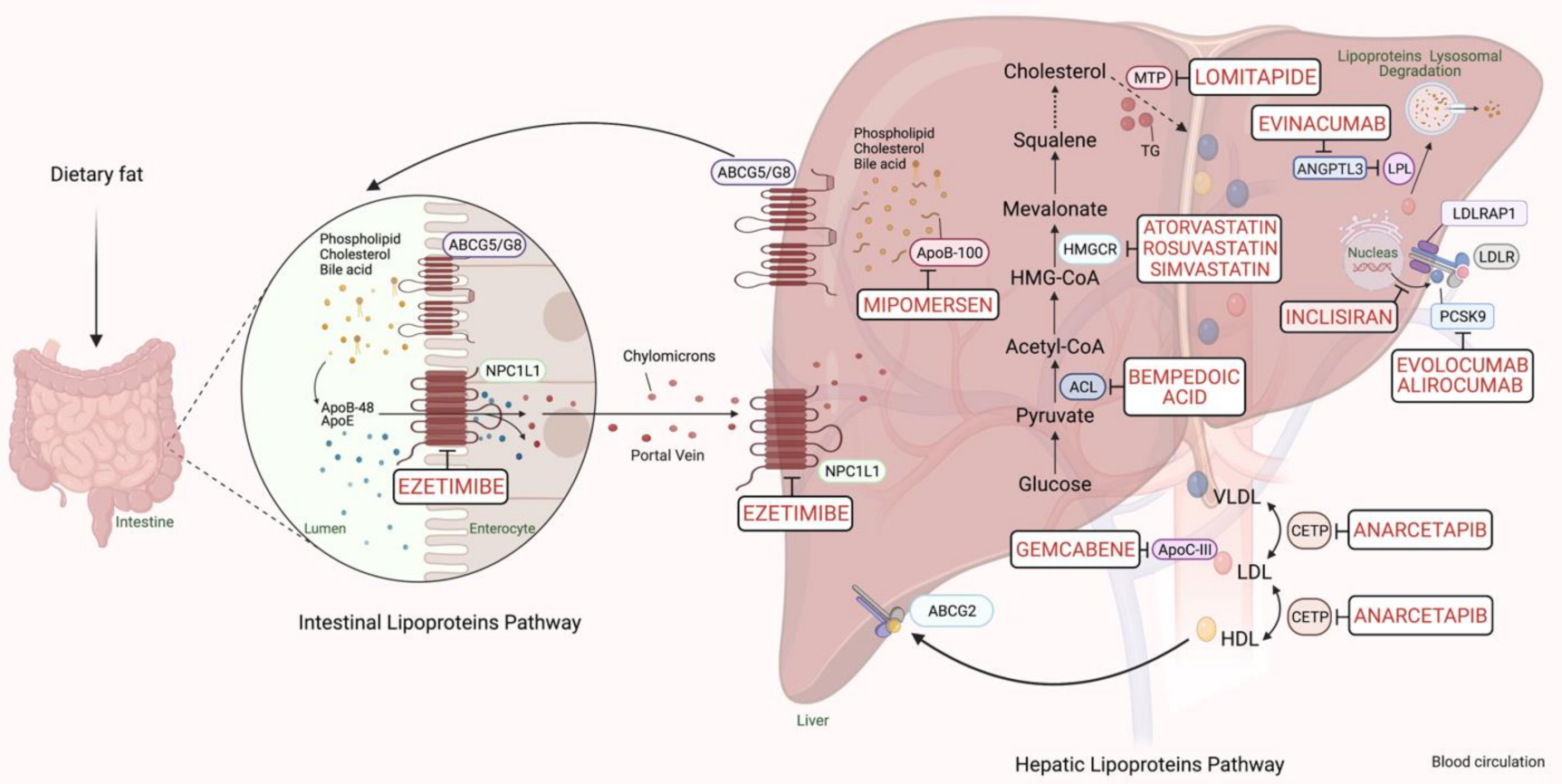
:1. Introduction
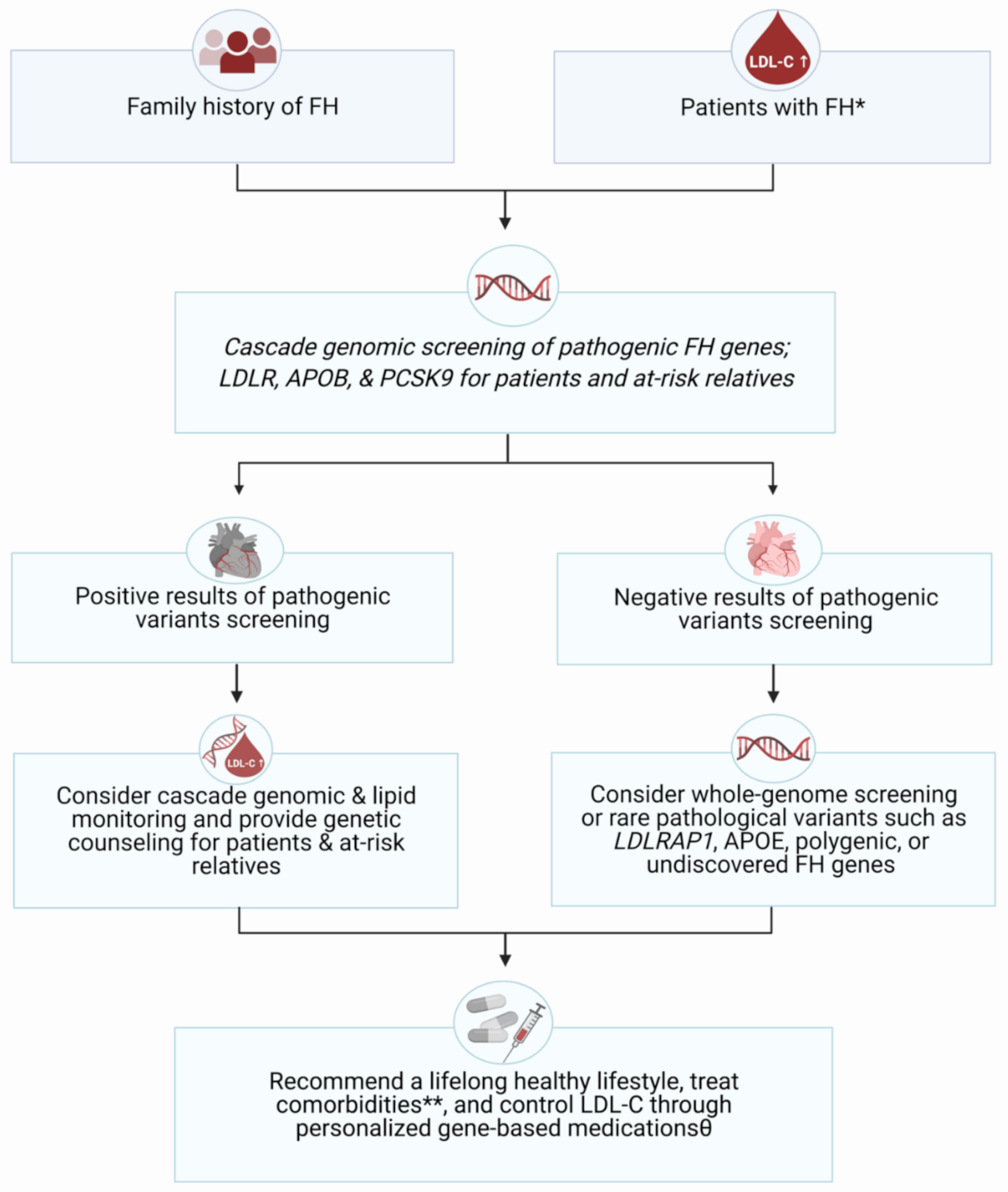
2. FH Management
3. Pharmacogenomics of Statin in FH
3.1. SNPs Linked to Pharmacodynamics of Statins in FH

{kind=link}
{kind=link}
{kind=link}
| Gene | Significant Mutation * | Patients | Population | Sample Size | Treatment and Daily Dose | Clinical Findings | Author, Year (References) |
|---|---|---|---|---|---|---|---|
| LDLR | FH1 (C206G) & FH2 (G408A) | Het-FH | Afrikaners | 20 | Simvastatin 40 mg | TC reduction is higher in patients with FH2 than FH1 | Jeenah et al., 1993 [19] |
| LDLR | C660X, D147H, & 652delGGT | Het-FH | Israeli | 64 | Fluvastatin 40 mg | Reduction of LDL-C, apoA, and elevation of HDL-C depend variously on LDLR mutations | Leitersdorf et al., 1993 [23] |
| APOE | E2,3, & 4 alleles | Het-FH | Canadian | 49 | Lovastatin 80 mg | Statin sensitive is higher in men with E4 than E3 or E2 or women with any APOE phenotype | Carmena et al., 1993 [35] |
| LDLR | FHTONAMI-1 (Del exon15) &FHKANAZAWA (C665T) | Het-FH | Japanese | 12 | Pravastatin & cholestyramine | LDL-C reduction is higher in patients with FHKANAZAWA than FH1 FHTONAMI-1 | Kajinami et al., 1998 [20] |
| LDLR | W66G, C646Y, & deletion>15 kb | Het-FH | Canadian | 63 | Simvastatin 20 mg | LDL-C reduction is higher in patients with C646Y & deletion > 15 kb than W66G | Couture et al., 1998 [21] |
| LDLR | Severe and mild LDLR | Het-FH | British | 42 | Simvastatin + bile acid sequestrant | LDL-C is higher in patients with severe than mild mutation | Sun et al., (1998) [33] |
| LDLR | Null and defective LDLR | Het-FH | British | 109 | Simvastatin | LDL-C reduction is higher in patients with defective than null mutation | Heath et al., (1999) [31] |
| LDLR | AvaII (rs5925T>C), HincII (rs688C>T), & PvuII (rs2569542A>G) | Het-FH | Brazilian | 55 | Fluvastatin 40–80 mg | LDL-C, TC, & ApoB reduction is higher in patients with AvaII & PvuII than HincII | Salazar et al., 2000 [22] |
| LDLR | Null and defective LDLR | FH | Spanish | 55 | Simvastatin 20 mg | Low HDL-C & poor statin response are higher in patients with defective than null mutations | Chaves et al., (2001) [32] |
| APOE | E4 allele | Het-FH | British | 19 | Atorvastatin 10 mg + bile acid sequestrant | Poor statins response is high in patients with E4 phenotype | O’Neill et al., 2001 [36] |
| LDLR | Null and defective LDLR | Het-FH | Canadian | 63 | Atorvastatin 20 mg | LDL-C reduction is higher in patients with null than defective mutation | Vohl et al., (2002) [37] |
| LDLR | G1775A, G1646A, & C858A | Het-FH | Greek | 49 | Atorvastatin 20 mg | LDL-C & ApoB reduction is higher in patients with G1775A than G1646A & C858A | Miltiadous et al., 2005 [24] |
| MTP | c.493 GT | Het-FH | Spanish | 222 | Atorvastatin 20 mg | High reduction of TG in men and low reduction of VLDL & TG in women with c.493 GT allele | García-Garc ía et al., 2005 [38] |
| CETP | −867 and Ex14/I405V | Het-FH | Israeli | 76 | Fluvastatin 40 mg | LDL-C reduction is high among CETP & MDR1 mutants | Bercovich et al., 2006 [39] |
| MDR1 | c.(G2677T) and c.(C3435T) | ||||||
| LDLR | Null and defective LDLR | Het-FH | Spanish | 811 | Simvastatin or atorvastatin 80 mg ± bile acid sequestrant | PCVD & TC is higher in patients with null than defective mutations | Alonso et al., 2008 [40] |
| ABCG2 | rs2231142 | FH | Chinese | 386 | Rosuvastatin 10 mg | High LDL-C reduction among patients with AA genotype | Hu et al., 2010 [41] |
| LDLR | Null and defective LDLR | FH | Spanish | 387 | Maximum statin doses ** + ezetimibe 10 mg | Poor LLT response & high PCVD in patients with null than defective mutations | Mata et al. (2011) [42] |
| LDLR | W556R | Twins with Hom-FH and parents with Het-FH (one family) | Turkish | 4 | Simvastatin 40 mg + ezetimibe 10 mg or LDL apheresi | Hom-FH have a low LDL-C reduction and high statin resistance, but Het-FH respond to statin with 60% LDL-C reduction | Schaefer et al., 2012 [43] |
| CYP3A4 | rs2740574 | FH | Chilean | 142 | Atorvastatin 10 mg | High statin sensitivity among patients with CYP3A4 mutations | Rosales et al., 2012 [44] |
| ANRIL | rs1333049 | FH with CVD | Pakistani | 611 | Atorvastatin 10, 20 or 40 mg | High LDL-C, TC, & TG reduction in patients with CC genotype | Ahmed et al., 2013 [45] |
| LDLR | Null (W66G) and defective (C646Y) LDLR | Het-FH | Brazilian | 156 | Atorvastatin 10, 20 or 40 mg | LDL-C reduction is more in patients with defective than with null mutation | Santos et al., 2014 [30] |
| POR | rs1057868 | FH | Greek | 105 | Atorvastatin 10, 20 and 40 mg | High LDL-C & TC reduction in patients with 1/1 genotype | Drogari et al., 2014 [46] |
| MYLIP | rs9370867 | Het-FH | Brazilian | 156 | Atorvastatin 10–80 mg ± ezetimibe 10 mg | High LDL-C reduction in patients with AA genotype | Santos et al., 2014 [47] |
| PSCK9 | E32K | Hom-FH | Japanese | 1055 | Atorvastatin 80 mg & ezetimibe 10 mg | PSCK9 gain-of-function variants significantly worsen LDLR phenotype and decrease LDL-C reduction | Mabuchi, et al., 2014 [48] |
| LDLR | Double allele | ||||||
| LDLR | Null and defective LDLR | FH | Spanish | 4132 | Maximum statin doses ** + ezetimibe 10 mg | Poor LLT response & CVD events are higher in null than in defective mutation | Perez de Isla et al., 2016 [14] |
| LDLR | p.(Cys155Gly) | Hom-FH | Belgian | 8 | Atorvastatin 80 mg, ezetimibe 10 mg, cholestyramine | LLT efficacy is attenuated in patients with nonsense LDLR mutations | Sanna et al., 2016 [34] |
| HMGCR | rs3846662 | Het-FH | French Canadian | 106 | Statin + LLTΩ | Poor statin response among HMGCR mutants | Leduc et al., 2016 [49] |
| LDLR | W87G, C368Y, T726I, G2fsX214, D47N, N97H, E101K, C216fsX, L582P, C667Y, & LDLR-17-18 del | Het-FH | American and Canadian | 139 | Atorvastatin 40/80 mg, rosuvastatin 20/40 mg or simvastatin 40/80 mg, + Bococizumab 0.25, 1, 3, or 6 mg/kg | Bococizumab effecacy is higher than statin in reducing LDL-C across LDLR & APOB variants | Fazio et al., 2018 [50] |
| APOB | R3527Q | ||||||
| LDLR | Het-LDLR mutation | FH | Spanish | 22 | Maximum statin doses ** ± ezetimibe 10 mg | LDL-C reduction is higher in patients with p.(Leu167del) mutation than LDLR | Bea et al., 2019 [51] |
| APOE | p.(Leu167del) | ||||||
| LDLR SLCO1B1 ABCB11 CYP3A5 | rs28941776 c.(521T>C; SLCO1B1*5) & c.(388A>G; SLCO1B1*1B) rs2287622 CYP3A5*3 | FH | Caucasian | 1 | Rosuvastatin 40 mg & ezetimibe 10 mg | Loss-of-function mutations enhance statin myotoxicity and delay its response | Dagli-Hernandez et al., 2021 [52] |
| LDLR | c.(2027delG), p. (Gly676Alafs*33) | FH (2 families) | Saudi | 12 | Statin + ezetimibe | Clinical manifestations and poor LLT response depend on LDLR variants | Awan et al., 2021 [29] |
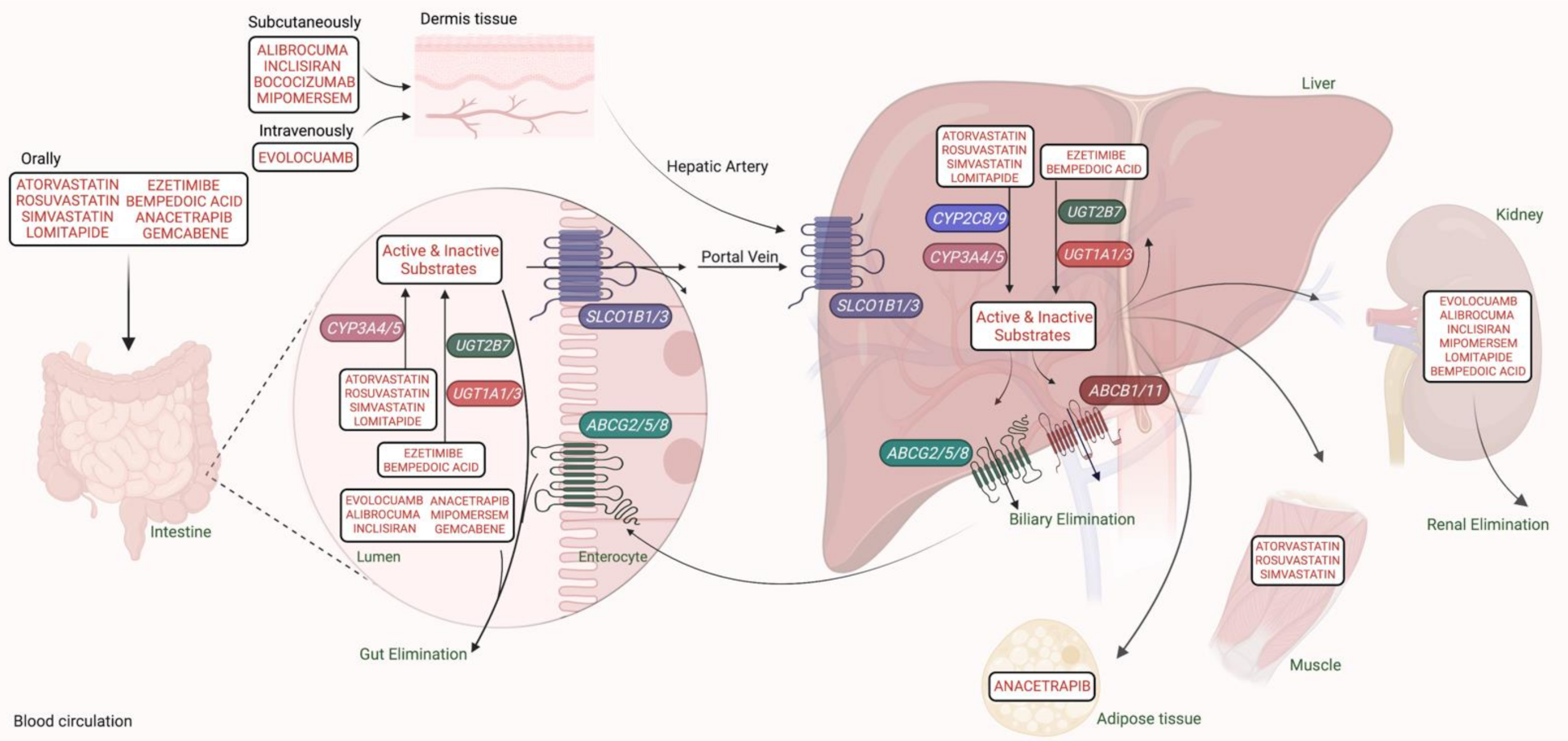
3.2. SNPs Linked to Pharmacokinetics and Pharmacotoxicity of Statins in FH
4. Pharmacogenomics of Non-Statin Lipid-Lowering Therapies in FH
4.1. Ezetimibe
| Gene | Significant Mutation * | Patients | Population | Sample Size | Treatment and Daily Dose | Clinical Findings | Author, Year (References) |
|---|---|---|---|---|---|---|---|
| Non-statin Lipid-Lowering Therapies | |||||||
| LDLR | Defective and negative LDLR | Hom-FH | South African | 8 | Evolocumab 140–420 mg every 2–4 weeks for 3 months | Evolocumab is reducing LDL-C in LDLR-defective but not in negative cases | Stein et al., 2013 [65] |
| LDLR | Defective and negative LDLR | Hom-FH | 10 countries ** | 50 | Evolocumab 420 mg every 4 weeks for 3 months | Evolocumab responses is LDLR-genotype dependent with higher sensitivity in LDLR-defective patients | Raal et al., 2015 [66] |
| PCSK9 | rs28942111 (S127R) rs28942112 (F216L) | Het-FH | 27 countries ** | 2341 | Statin maximum dose + LLT & alirocumab 150 mg/2 weeks for 78 weeks | Alirocumab is significantly reducing LDL-C in PCSK9 gain-of-function variants | Robinson et al., 2015 [67] |
| LDLR | c.(1646G <A) | Hom-FH | Italian | 15 | Simvastatin 40 mg, ezetimibe 10 mg, & lomitapide 5–60 mg | Lomitapide is significantly and safely decreasing the cholesterol levels | D’Erasmo et al., 2017 [68] |
| LDLRAP1 | c.(432_433insA) | ||||||
| LDLR | Defective and negative LDLR | Hom-FH | South African | 22 | Mivastatin and evolocumab | Evolocumab is effective in defective- and not in negative-LDLR variants | Thedrez et al., 2017 [15] |
| APOB | R3500Q (rs5742904) | Het-FH | Caucasian | 1 | Atorvastatin 80 mg, ezetimibe 10 mg, lomitapide, & evolocumab 140 mg | ApoB defect is enhancing LDL-C reduction | Andersen et al., 2017 [69] |
| LDLRAP1 | c.136 C > T (406) | AR-FH | German | 1 | Atorvastatin 80 mg, ezetimibe 10 mg, lomitapide, & evolocumab 140 mg | Evolocumab is reducing LDL-C by 37% among LDLRAP1 mutants | Fahy et al., 2017 [70] |
| LDLR | Two null alleles | Hom-FH | American | 9 | LLTΩ + Evolocumab 420 mg/4 weeks | Evinacumab is controlling cholesterol independently of LDLR variants | Gaudet et al., 2017 [71] |
| LDLR | c.2043C.A, p. (Cys 681A) | Het-FH | Lebanese American | 1 | Rosuvastatin, ezetimibe, & evolocumab 140 mg/2 weeks for 2 months, then alirocumab 150 mg/ 2 weeks | Alirocumab efficacy is higher than evolocumab & standard LLT | Doyle et al., 2018 [72] |
| LDLR | p.(Thr434Arg) | Hom-FH | Spanish | 2 | LLTΩ & lomitapide 20–40 mg | Lomitapide is potent and safe as adjunct therapy | Real et al., 2018 [73] |
| LDLRAP1 | c.1A > G | AR-FH | Spanish | 3 | Atorvastatin, ezetimibe, & evolocumab | Evolocumab effecacy is lower in LDLR & LDLRAP1 variants | Rodríguez-Jiménez et al., 2019 [74] |
| LDLR | p.(Cys352Ser) & p.(Asn825Lys) | ||||||
| LDLR PCSK9 APOB | p.(Trp87Gly), p.(Gln254Pro), & p.(Ala627Profs*38) | Hom-FH | Chinese | 9 | LLTΩ + evinacumab 250 mg | Evinacumab is controlling cholesterol independently of LDLR variants | Banerjee et al., 2019 [75] |
| PSCK9 | c.137 G>T, p.(Arg46Leu) | Hom-FH | Caucasian | 3 | LLTΩ + Evolocumab 420 mg/ 4 weeks | Evolocumab is strongly reducing LDL-C and CVD in PSCK9 loss-of-function mutants | Bayonaet al., 2020 [76] |
| LDLR | c. 902A>G, p.(Asp301Gly) | ||||||
| LDLR | p.(Pro685Leu) | Hom-FH | Indian | 1 | LLTΩ & evolocumab 420 mg/4 weeks + lomitapide 5–60 mg | Lomitapide is powerfully reducing lipid profile and CVD risk | Velvet et al., 2020 [77] |
| LDLR | Null mutation in both alleles | Hom-FH | 13 countries ** | 69 | Atorvastatin 80 mg, ezetimibe 10 mg, lomitapide, & alirocumab 150 mg/2 weeks for 12 weeks | Alirocumab is effective in controlling the lipid profile | Blom et al., 2020 [78] |
| LDLR | c.2027delG, p.(G676Afs*33) | Hom-FH | Saudi | 2 | Rosuvastatin 40 mg, ezetimibe 10 mg, evolocumab 420 mg/month, & lomitapide 5–40 mg | Lomitapide is robustly reducing cholesterol and CVD events | Mahzari et al., 2021 [79] |
| Novel Lipid-Lowering Therapies | |||||||
| LDLR APOB PCSK9 | Deficient and defective | Het-FH | 9 countries *** | 306 | LLTΩ & anacetrapib 100 mg for 12 months | Anacetrapib is substantially reducing LDL-C across all genotypes | Kastelein et al., 2016 [80] |
| LDLR | Defective and negative LDLR | Hom-FH | American | 8 | Statins, ezetimibe, mipomersen, lomitapide, PCSK9 inhibitors, & gemcabene 300, 600 or 900 mg | Gemcabene is reducing LDL-C in uncontrolled cases under LLT treatment | Gaudet et al., 2019 [81] |
| LDLR PCSK9 APOB LDLRAP1 | Pathogenic causative Gain-of-function Pathogenic Pathogenic | Het-FH | 8 countries ** | 432 | LLTΩ & inclisiran 300 mg/3 months | Inclisiran is significantly reducing LDL- C in LDLR variants | Raal et al., 2020 [82] |
| LDLR APOB | Pathogenic | Het-FH | Various countries ** | 1887 | LLTΩ, evolocumab or alirocumab, & inclisiran | PCSK9 inhibitors is reducing LDL-C among all genetic mutations | Brandts et al., 2021 [83] |
4.2. Monoclonal Antibodies to PCSK9
4.3. Mipomersen
4.4. Lomitapide
5. Pharmacogenomics of Novel Lipid-Lowering Therapies in FH
5.1. Evinacumab
5.2. Bempedoic Acid
5.3. Gemcabene
5.4. CETP Inhibitor
6. Conclusions and Clinical Prospect of the Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Khachadurian, A.K. The Inheritance of Essential Familial Hypercholesterolemia. Am. J. Med. 1964, 37, 402–407. [Google Scholar] [CrossRef]
- Lui, D.T.W.; Lee, A.C.H.; Tan, K.C.B. Management of Familial Hypercholesterolemia: Current Status and Future Perspectives. J. Endocr. Soc. 2021, 5, bvaa122. [Google Scholar] [CrossRef] [PubMed]
- Hayat, M.; Kerr, R.; Bentley, A.R.; Rotimi, C.N.; Raal, F.J.; Ramsay, M. Genetic associations between serum low LDL-cholesterol levels and variants in LDLR, APOB, PCSK9 and LDLRAP1 in African populations. PLoS ONE 2020, 15, e0229098, Correction in 2021, 16, e0249478. [Google Scholar] [CrossRef]
- Fahed, A.C.; Nemer, G.M. Familial hypercholesterolemia: The lipids or the genes? Nutr. Metab. 2011, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Galema-Boers, A.M.; Lenzen, M.J.; Engelkes, S.R.; Sijbrands, E.J.; Roeters van Lennep, J.E. Cardiovascular risk in patients with familial hypercholesterolemia using optimal lipid-lowering therapy. J Clin. Lipidol. 2018, 12, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Corrigendum to: 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 4255. [CrossRef] [PubMed] [Green Version]
- Kamar, A.; Khalil, A.; Nemer, G. The Digenic Causality in Familial Hypercholesterolemia: Revising the Genotype-Phenotype Correlations of the Disease. Front. Genet. 2020, 11, 572045. [Google Scholar] [CrossRef]
- Borges, J.B.; Oliveira, V.F.; Ferreira, G.M.; Los, B.; Barbosa, T.; Marcal, E.; Dagli-Hernandez, C.; de Freitas, R.C.C.; Bortolin, R.H.; Mori, A.A.; et al. Genomics, epigenomics and pharmacogenomics of familial hypercholesterolemia (FHBGEP): A study protocol. Res. Soc. Adm. Pharm. 2021, 17, 1347–1355. [Google Scholar] [CrossRef]
- Chahine, J.; Kreykes, S.; Van’t Hof, J.R.; Duprez, D.; Nijjar, P. Variable and Severe Phenotypic Expression of the “Lebanese Allele” in Two Sisters with Familial Hypercholesterolemia. Vasc. Health Risk Manag. 2021, 17, 415–419. [Google Scholar] [CrossRef]
- Fahed, A.C.; Bitar, F.F.; Khalaf, R.I.; Moubarak, E.M.; Azar, S.T.; Nemer, G.M. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine 2012, 42, 445–448. [Google Scholar] [CrossRef]
- Hartgers, M.L.; Besseling, J.; Stroes, E.S.; Wittekoek, J.; Rutten, J.H.W.; de Graaf, J.; Visseren, F.L.J.; Imholz, B.P.M.; Roeters van Lennep, J.E.; Huijgen, R.; et al. Achieved LDL cholesterol levels in patients with heterozygous familial hypercholesterolemia: A model that explores the efficacy of conventional and novel lipid-lowering therapy. J. Clin. Lipidol. 2018, 12, 972–980.e1. [Google Scholar] [CrossRef]
- Marziliano, N.; Medoro, A.; Mignogna, D.; Saccon, G.; Folzani, S.; Reverberi, C.; Russo, C.; Intrieri, M. Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection. Diagnostics 2021, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Luirink, I.K.; Wiegman, A.; Kusters, D.M.; Hof, M.H.; Groothoff, J.W.; de Groot, E.; Kastelein, J.J.P.; Hutten, B.A. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 2019, 381, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Perez de Isla, L.; Alonso, R.; Watts, G.F.; Mata, N.; Saltijeral Cerezo, A.; Muniz, O.; Fuentes, F.; Diaz-Diaz, J.L.; de Andres, R.; Zambon, D.; et al. Attainment of LDL-Cholesterol Treatment Goals in Patients With Familial Hypercholesterolemia: 5-Year SAFEHEART Registry Follow-Up. J. Am. Coll. Cardiol. 2016, 67, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Thedrez, A.; Blom, D.J.; Ramin-Mangata, S.; Blanchard, V.; Croyal, M.; Chemello, K.; Nativel, B.; Pichelin, M.; Cariou, B.; Bourane, S.; et al. Homozygous Familial Hypercholesterolemia Patients With Identical Mutations Variably Express the LDLR (Low-Density Lipoprotein Receptor): Implications for the Efficacy of Evolocumab. Arter. Thromb. Vasc. Biol. 2018, 38, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Fahed, A.C.; El-Hage-Sleiman, A.K.; Farhat, T.I.; Nemer, G.M. Diet, genetics, and disease: A focus on the middle East and north Africa region. J. Nutr. Metab. 2012, 2012, 109037. [Google Scholar] [CrossRef]
- Ramaswami, U.; Futema, M.; Bogsrud, M.P.; Holven, K.B.; Roeters van Lennep, J.; Wiegman, A.; Descamps, O.S.; Vrablik, M.; Freiberger, T.; Dieplinger, H.; et al. Comparison of the characteristics at diagnosis and treatment of children with heterozygous familial hypercholesterolaemia (FH) from eight European countries. Atherosclerosis 2020, 292, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.S.; Psaty, B.M.; et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin. Pharm. Ther. 2019, 106, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeenah, M.; September, W.; Graadt van Roggen, F.; de Villiers, W.; Seftel, H.; Marais, D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia. Atherosclerosis 1993, 98, 51–58. [Google Scholar] [CrossRef]
- Kajinami, K.; Yagi, K.; Higashikata, T.; Inazu, A.; Koizumi, J.; Mabuchi, H. Low-density lipoprotein receptor genotype-dependent response to cholesterol lowering by combined pravastatin and cholestyramine in familial hypercholesterolemia. Am. J. Cardiol. 1998, 82, 113–117. [Google Scholar] [CrossRef]
- Couture, P.; Brun, L.D.; Szots, F.; Lelievre, M.; Gaudet, D.; Despres, J.P.; Simard, J.; Lupien, P.J.; Gagne, C. Association of specific LDL receptor gene mutations with differential plasma lipoprotein response to simvastatin in young French Canadians with heterozygous familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 1998, 18, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Salazar, L.A.; Cavalli, S.A.; Hirata, M.H.; Diament, J.; Forti, N.; Giannini, S.D.; Nakandakare, E.R.; Bertolami, M.C.; Hirata, R.D. Polymorphisms of the low-density lipoprotein receptor gene in Brazilian individuals with heterozygous familial hypercholesterolemia. Braz. J. Med. Biol. Res. 2000, 33, 1301–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitersdorf, E.; Eisenberg, S.; Eliav, O.; Friedlander, Y.; Berkman, N.; Dann, E.J.; Landsberger, D.; Sehayek, E.; Meiner, V.; Wurm, M.; et al. Genetic determinants of responsiveness to the HMG-CoA reductase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation 1993, 87, III35-44. [Google Scholar]
- Miltiadous, G.; Xenophontos, S.; Bairaktari, E.; Ganotakis, M.; Cariolou, M.; Elisaf, M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharm. Genom. 2005, 15, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 2008, 200, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muallem, H.; North, K.E.; Kakoki, M.; Wojczynski, M.K.; Li, X.; Grove, M.; Boerwinkle, E.; Wilhelmsen, K.C.; Heiss, G.; Maeda, N. Quantitative effects of common genetic variations in the 3′UTR of the human LDL-receptor gene and their associations with plasma lipid levels in the Atherosclerosis Risk in Communities study. Hum. Genet. 2007, 121, 421–431. [Google Scholar] [CrossRef]
- Mangravite, L.M.; Medina, M.W.; Cui, J.; Pressman, S.; Smith, J.D.; Rieder, M.J.; Guo, X.; Nickerson, D.A.; Rotter, J.I.; Krauss, R.M. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arter. Thromb. Vasc. Biol. 2010, 30, 1485–1492. [Google Scholar] [CrossRef] [Green Version]
- Lahoz, C.; Pena, R.; Mostaza, J.M.; Laguna, F.; Garcia-Iglesias, M.F.; Taboada, M.; Pinto, X. Baseline levels of low-density lipoprotein cholesterol and lipoprotein (a) and the AvaII polymorphism of the low-density lipoprotein receptor gene influence the response of low-density lipoprotein cholesterol to pravastatin treatment. Metabolism 2005, 54, 741–747. [Google Scholar] [CrossRef]
- Awan, Z.A.; Rashidi, O.M.; Al-Shehri, B.A.; Jamil, K.; Elango, R.; Al-Aama, J.Y.; Hegele, R.A.; Banaganapalli, B.; Shaik, N.A. Saudi Familial Hypercholesterolemia Patients With Rare LDLR Stop Gain Variant Showed Variable Clinical Phenotype and Resistance to Multiple Drug Regimen. Front. Med. 2021, 8, 694668. [Google Scholar] [CrossRef]
- Santos, P.C.; Morgan, A.C.; Jannes, C.E.; Turolla, L.; Krieger, J.E.; Santos, R.D.; Pereira, A.C. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2014, 233, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Heath, K.E.; Gudnason, V.; Humphries, S.E.; Seed, M. The type of mutation in the low density lipoprotein receptor gene influences the cholesterol-lowering response of the HMG-CoA reductase inhibitor simvastatin in patients with heterozygous familial hypercholesterolaemia. Atherosclerosis 1999, 143, 41–54. [Google Scholar] [CrossRef]
- Chaves, F.J.; Real, J.T.; Garcia-Garcia, A.B.; Civera, M.; Armengod, M.E.; Ascaso, J.F.; Carmena, R. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: Influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J. Clin. Endocrinol. Metab. 2001, 86, 4926–4932. [Google Scholar] [CrossRef]
- Sun, X.M.; Patel, D.D.; Knight, B.L.; Soutar, A.K. Influence of genotype at the low density lipoprotein (LDL) receptor gene locus on the clinical phenotype and response to lipid-lowering drug therapy in heterozygous familial hypercholesterolaemia. The Familial Hypercholesterolaemia Regression Study Group. Atherosclerosis 1998, 136, 175–185. [Google Scholar] [CrossRef]
- Sanna, C.; Stephenne, X.; Revencu, N.; Smets, F.; Sassolas, A.; Di Filippo, M.; Descamps, O.S.; Sokal, E.M. Homozygous familial hypercholesterolemia in childhood: Genotype-phenotype description, established therapies and perspectives. Atherosclerosis 2016, 247, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Carmena, R.; Roederer, G.; Mailloux, H.; Lussier-Cacan, S.; Davignon, J. The response to lovastatin treatment in patients with heterozygous familial hypercholesterolemia is modulated by apolipoprotein E polymorphism. Metabolism 1993, 42, 895–901. [Google Scholar] [CrossRef]
- O’Neill, F.H.; Patel, D.D.; Knight, B.L.; Neuwirth, C.K.; Bourbon, M.; Soutar, A.K.; Taylor, G.W.; Thompson, G.R.; Naoumova, R.P. Determinants of variable response to statin treatment in patients with refractory familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2001, 21, 832–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vohl, M.C.; Szots, F.; Lelievre, M.; Lupien, P.J.; Bergeron, J.; Gagne, C.; Couture, P. Influence of LDL receptor gene mutation and apo E polymorphism on lipoprotein response to simvastatin treatment among adolescents with heterozygous familial hypercholesterolemia. Atherosclerosis 2002, 160, 361–368. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.B.; Gonzalez, C.; Real, J.T.; Martin de Llano, J.J.; Gonzalez-Albert, V.; Civera, M.; Chaves, F.J.; Ascaso, J.F.; Carmena, R. Influence of microsomal triglyceride transfer protein promoter polymorphism -493 GT on fasting plasma triglyceride values and interaction with treatment response to atorvastatin in subjects with heterozygous familial hypercholesterolaemia. Pharm. Genom. 2005, 15, 211–218. [Google Scholar] [CrossRef]
- Bercovich, D.; Friedlander, Y.; Korem, S.; Houminer, A.; Hoffman, A.; Kleinberg, L.; Shochat, C.; Leitersdorf, E.; Meiner, V. The association of common SNPs and haplotypes in the CETP and MDR1 genes with lipids response to fluvastatin in familial hypercholesterolemia. Atherosclerosis 2006, 185, 97–107. [Google Scholar] [CrossRef]
- Alonso, R.; Mata, N.; Castillo, S.; Fuentes, F.; Saenz, P.; Muniz, O.; Galiana, J.; Figueras, R.; Diaz, J.L.; Gomez-Enterria, P.; et al. Cardiovascular disease in familial hypercholesterolaemia: Influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008, 200, 315–321. [Google Scholar] [CrossRef]
- Hu, M.; Lui, S.S.; Mak, V.W.; Chu, T.T.; Lee, V.W.; Poon, E.W.; Tsui, T.K.; Ko, G.T.; Baum, L.; Tam, L.S.; et al. Pharmacogenetic analysis of lipid responses to rosuvastatin in Chinese patients. Pharm. Genom. 2010, 20, 634–637. [Google Scholar] [CrossRef]
- Mata, N.; Alonso, R.; Badimon, L.; Padro, T.; Fuentes, F.; Muniz, O.; Perez-Jimenez, F.; Lopez-Miranda, J.; Diaz, J.L.; Vidal, J.I.; et al. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis 2011, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, J.R.; Kurt, B.; Sattler, A.; Klaus, G.; Soufi, M. Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R). Clin. Res. Cardiol. Suppl. 2012, 7, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, A.; Alvear, M.; Cuevas, A.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Identification of pharmacogenetic predictors of lipid-lowering response to atorvastatin in Chilean subjects with hypercholesterolemia. Clin. Chim. Acta 2012, 413, 495–501. [Google Scholar] [CrossRef]
- Ahmed, W.; Ali, I.S.; Riaz, M.; Younas, A.; Sadeque, A.; Niazi, A.K.; Niazi, S.H.; Ali, S.H.; Azam, M.; Qamar, R. Association of ANRIL polymorphism (rs1333049:C>G) with myocardial infarction and its pharmacogenomic role in hypercholesterolemia. Gene 2013, 515, 416–420. [Google Scholar] [CrossRef]
- Drogari, E.; Ragia, G.; Mollaki, V.; Elens, L.; Van Schaik, R.H.; Manolopoulos, V.G. POR*28 SNP is associated with lipid response to atorvastatin in children and adolescents with familial hypercholesterolemia. Pharmacogenomics 2014, 15, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Morgan, A.C.; Jannes, C.E.; Krieger, J.E.; Santos, R.D.; Pereira, A.C. The MYLIP p.N342S polymorphism is associated with response to lipid-lowering therapy in Brazilian patients with familial hypercholesterolemia. Pharm. Genom. 2014, 24, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, H.; Nohara, A.; Noguchi, T.; Kobayashi, J.; Kawashiri, M.A.; Inoue, T.; Mori, M.; Tada, H.; Nakanishi, C.; Yagi, K.; et al. Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis 2014, 236, 54–61. [Google Scholar] [CrossRef]
- Leduc, V.; Bourque, L.; Poirier, J.; Dufour, R. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharm. Genom. 2016, 26, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fazio, S.; Robertson, D.G.; Joh, T.; Wan, H.; Riel, T.; Forgues, P.; Baum, C.M.; Garzone, P.D.; Gumbiner, B. Effects of 12 weeks of treatment with intravenously administered bococizumab, a humanized monoclonal antibody blocking proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects on high-dose statin. Cardiovasc Ther. 2018, 36, e12308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bea, A.M.; Lamiquiz-Moneo, I.; Marco-Benedi, V.; Mateo-Gallego, R.; Perez-Calahorra, S.; Jarauta, E.; Martin, C.; Cenarro, A.; Civeira, F. Lipid-lowering response in subjects with the p.(Leu167del) mutation in the APOE gene. Atherosclerosis 2019, 282, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagli-Hernandez, C.; de Freitas, R.C.C.; Marcal, E.; Goncalves, R.M.; Faludi, A.A.; Borges, J.B.; Bastos, G.M.; Los, B.; Mori, A.A.; Bortolin, R.H.; et al. Late response to rosuvastatin and statin-related myalgia due to SLCO1B1, SLCO1B3, ABCB11, and CYP3A5 variants in a patient with Familial Hypercholesterolemia: A case report. Ann. Transl. Med. 2021, 9, 76. [Google Scholar] [CrossRef]
- Charland, S.L.; Agatep, B.C.; Herrera, V.; Schrader, B.; Frueh, F.W.; Ryvkin, M.; Shabbeer, J.; Devlin, J.J.; Superko, H.R.; Stanek, E.J. Providing patients with pharmacogenetic test results affects adherence to statin therapy: Results of the Additional KIF6 Risk Offers Better Adherence to Statins (AKROBATS) trial. Pharm. J. 2014, 14, 272–280. [Google Scholar] [CrossRef]
- Paquette, M.; Bernard, S.; Thanassoulis, G.; Baass, A. LPA genotype is associated with premature cardiovascular disease in familial hypercholesterolemia. J. Clin. Lipidol. 2019, 13, 627–633.e1. [Google Scholar] [CrossRef]
- Chasman, D.I.; Giulianini, F.; MacFadyen, J.; Barratt, B.J.; Nyberg, F.; Ridker, P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.F.; Hyde, C.L.; Wood, L.S.; Paciga, S.A.; Hinds, D.A.; Cox, D.R.; Hovingh, G.K.; Kastelein, J.J. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ. Cardiovasc. Genet. 2009, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- de Keyser, C.E.; Becker, M.L.; Hofman, A.; Lous, J.J.; Uitterlinden, A.G.; Visser, L.E.; Stricker, B.H. The rs13064411 polymorphism in the WDR52 gene, associated with PCSK9 levels, modifies statin-induced changes in serum total and LDL cholesterol levels. Pharm. Genom. 2015, 25, 134–142. [Google Scholar] [CrossRef]
- To, K.K.; Hu, M.; Tomlinson, B. Expression and activity of ABCG2, but not ABCB1 or OATP1B1, are associated with cholesterol levels: Evidence from in vitro and in vivo experiments. Pharmacogenomics 2014, 15, 1091–1104. [Google Scholar] [CrossRef]
- Mladenovska, K.; Grapci, A.D.; Vavlukis, M.; Kapedanovska, A.; Eftimov, A.; Geshkovska, N.M.; Nebija, D.; Dimovski, A.J. Influence of SLCO1B1 polymorphisms on atorvastatin efficacy and safety in Macedonian subjects. Pharmazie 2017, 72, 288–295. [Google Scholar] [CrossRef]
- Leon-Cachon, R.B.R.; Bamford, A.D.; Meester, I.; Barrera-Saldana, H.A.; Gomez-Silva, M.; Bustos, M.F.G. The atorvastatin metabolic phenotype shift is influenced by interaction of drug-transporter polymorphisms in Mexican population: Results of a randomized trial. Sci. Rep. 2020, 10, 8900. [Google Scholar] [CrossRef]
- Wendt, F.R.; Koller, D.; Pathak, G.A.; Jacoby, D.; Miller, E.J.; Polimanti, R. Biobank Scale Pharmacogenomics Informs the Genetic Underpinnings of Simvastatin Use. Clin. Pharm. Ther. 2021, 110, 777–785. [Google Scholar] [CrossRef]
- Berthold, H.K.; Laaksonen, R.; Lehtimaki, T.; Gylling, H.; Krone, W.; Gouni-Berthold, I. SREBP-1c gene polymorphism is associated with increased inhibition of cholesterol-absorption in response to ezetimibe treatment. Exp. Clin. Endocrinol. Diabetes 2008, 116, 262–267. [Google Scholar] [CrossRef]
- Ference, B.A.; Majeed, F.; Penumetcha, R.; Flack, J.M.; Brook, R.D. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: A 2 x 2 factorial Mendelian randomization study. J. Am. Coll. Cardiol. 2015, 65, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Komiya, C.; Shimizu, H.; Mishima, H.; Shiba, K.; Tsujimoto, K.; Ikeda, K.; Kashimada, K.; Dateki, S.; Yoshiura, K.I.; et al. A case of ezetimibe-effective hypercholesterolemia with a novel heterozygous variant in ABCG5. Endocr. J. 2020, 67, 1099–1105. [Google Scholar] [CrossRef]
- Stein, E.A.; Honarpour, N.; Wasserman, S.M.; Xu, F.; Scott, R.; Raal, F.J. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation 2013, 128, 2113–2120. [Google Scholar] [CrossRef]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; Stein, E.A.; Investigators, T. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Cefalu, A.B.; Noto, D.; Giammanco, A.; Averna, M.; Pintus, P.; Medde, P.; Vigna, G.B.; Sirtori, C.; Calabresi, L.; et al. Efficacy of Lomitapide in the Treatment of Familial Homozygous Hypercholesterolemia: Results of a Real-World Clinical Experience in Italy. Adv. Ther. 2017, 34, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.; Davis, T.; Testa, H.; Andersen, R.L. PCSK9 inhibitor therapy in homozygous familial defective apolipoprotein B-100 due to APOB R3500Q: A case report. J. Clin. Lipidol. 2017, 11, 1471–1474. [Google Scholar] [CrossRef]
- Fahy, E.F.; McCarthy, E.; Steinhagen-Thiessen, E.; Vaughan, C.J. A case of autosomal recessive hypercholesterolemia responsive to proprotein convertase subtilisin/kexin 9 inhibition. J. Clin. Lipidol. 2017, 11, 287–288. [Google Scholar] [CrossRef]
- Gaudet, D.; Gipe, D.A.; Pordy, R.; Ahmad, Z.; Cuchel, M.; Shah, P.K.; Chyu, K.Y.; Sasiela, W.J.; Chan, K.C.; Brisson, D.; et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2017, 377, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M. Differential Responses to the PCSK9 Inhibitors, Evolocumab and Alirocumab, in a Patient with Heterozygous Familial Hypercholesterolemia: A Case Report. Clin. Lipidol. 2018, 12, 558–559. [Google Scholar] [CrossRef]
- Real, J.; Arbona, C.; Goterris, R.; Ascaso, J.F. Management of homozygous familial hypercholesterolaemia in two brothers. BMJ Case Rep. 2018, 2018, bcr-2017. [Google Scholar] [CrossRef]
- Rodriguez-Jimenez, C.; Gomez-Coronado, D.; Frias Vargas, M.; Cerrato, F.; Lahoz, C.; Saban-Ruiz, J.; Gonzalez-Nieto, D.; Lasuncion, M.A.; Mostaza, J.M.; Rodriguez-Novoa, S. A new variant (c.1A>G) in LDLRAP1 causing autosomal recessive hypercholesterolemia: Characterization of the defect and response to PCSK9 inhibition. Atherosclerosis 2019, 284, 223–229. [Google Scholar] [CrossRef]
- Banerjee, P.; Chan, K.C.; Tarabocchia, M.; Benito-Vicente, A.; Alves, A.C.; Uribe, K.B.; Bourbon, M.; Skiba, P.J.; Pordy, R.; Gipe, D.A.; et al. Functional Analysis of LDLR (Low-Density Lipoprotein Receptor) Variants in Patient Lymphocytes to Assess the Effect of Evinacumab in Homozygous Familial Hypercholesterolemia Patients With a Spectrum of LDLR Activity. Arter. Thromb. Vasc. Biol. 2019, 39, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Bayona, A.; Arrieta, F.; Rodriguez-Jimenez, C.; Cerrato, F.; Rodriguez-Novoa, S.; Fernandez-Lucas, M.; Gomez-Coronado, D.; Mata, P. Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: A case report. Medicine 2020, 99, e21754. [Google Scholar] [CrossRef]
- Velvet, A.J.J.; Soran, H.; Clarke, B.; Motwani, M.; Ordoubadi, F.F.; Daniels, M.J. Homozygous familial hypercholesterolemia with an update on cholesterol management. Oxf. Med. Case Rep. 2020, 2020, omaa072. [Google Scholar] [CrossRef]
- Blom, D.J.; Harada-Shiba, M.; Rubba, P.; Gaudet, D.; Kastelein, J.J.P.; Charng, M.J.; Pordy, R.; Donahue, S.; Ali, S.; Dong, Y.; et al. Efficacy and Safety of Alirocumab in Adults With Homozygous Familial Hypercholesterolemia: The ODYSSEY HoFH Trial. J. Am. Coll. Cardiol. 2020, 76, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mahzari, M.; Zarif, H. Homozygous Familial Hypercholesterolemia (HoFH) in Saudi Arabia and Two Cases of Lomitapide Use in a Real-World Setting. Adv. Ther. 2021, 38, 2159–2169. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Besseling, J.; Shah, S.; Bergeron, J.; Langslet, G.; Hovingh, G.K.; Al-Saady, N.; Koeijvoets, M.; Hunter, J.; Johnson-Levonas, A.O.; et al. Anacetrapib as lipid-modifying therapy in patients with heterozygous familial hypercholesterolaemia (REALIZE): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet 2015, 385, 2153–2161. [Google Scholar] [CrossRef]
- Gaudet, D.; Durst, R.; Lepor, N.; Bakker-Arkema, R.; Bisgaier, C.; Masson, L.; Golden, L.; Kastelein, J.J.; Hegele, R.A.; Stein, E. Usefulness of Gemcabene in Homozygous Familial Hypercholesterolemia (from COBALT-1). Am. J. Cardiol. 2019, 124, 1876–1880. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Brandts, J.; Dharmayat, K.I.; Vallejo-Vaz, A.J.; Azar Sharabiani, M.T.; Jones, R.; Kastelein, J.J.P.; Raal, F.J.; Ray, K.K. A meta-analysis of medications directed against PCSK9 in familial hypercholesterolemia. Atherosclerosis 2021, 325, 46–56. [Google Scholar] [CrossRef]
- Naoumova, R.P.; Tosi, I.; Patel, D.; Neuwirth, C.; Horswell, S.D.; Marais, A.D.; van Heyningen, C.; Soutar, A.K. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: Long-term follow-up and treatment response. Arter. Thromb. Vasc. Biol. 2005, 25, 2654–2660. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.N.; Krempf, M.; Bruckert, E.; Donahue, S.; Yang, F.; Zhang, Y.; DiCioccio, A.T. Pharmacokinetic and pharmacodynamic assessment of alirocumab in patients with familial hypercholesterolemia associated with proprotein convertase subtilisin/kexin type 9 gain-of-function or apolipoprotein B loss-of-function mutations. J. Clin. Lipidol. 2019, 13, 970–978. [Google Scholar] [CrossRef] [Green Version]
- Reeskamp, L.F.; Kastelein, J.J.P.; Moriarty, P.M.; Duell, P.B.; Catapano, A.L.; Santos, R.D.; Ballantyne, C.M. Safety and efficacy of mipomersen in patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2019, 280, 109–117. [Google Scholar] [CrossRef]
- Raal, F.J.; Braamskamp, M.J.; Selvey, S.L.; Sensinger, C.H.; Kastelein, J.J. Pediatric experience with mipomersen as adjunctive therapy for homozygous familial hypercholesterolemia. J. Clin. Lipidol. 2016, 10, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Bjorn, L.; Leren, T.P.; Ose, L.; Hamsten, A.; Karpe, F. A functional polymorphism in the promoter region of the microsomal triglyceride transfer protein (MTP -493G/T) influences lipoprotein phenotype in familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2000, 20, 1784–1788. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Muthuramu, I.; Somanathan, S.; Zhang, H.; Bell, P.; He, Z.; Yu, H.; Zhu, Y.; Tretiakova, A.P.; Wilson, J.M. Developing a second-generation clinical candidate AAV vector for gene therapy of familial hypercholesterolemia. Mol. Ther. Methods Clin. Dev. 2021, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, B.M. Evinacumab Approval Adds a New Option for Homozygous Familial Hypercholesterolemia with a Hefty Price Tag. Circulation 2021, 143, 2494–2496. [Google Scholar] [CrossRef] [PubMed]
- Rubino, J.; MacDougall, D.E.; Sterling, L.R.; Hanselman, J.C.; Nicholls, S.J. Combination of bempedoic acid, ezetimibe, and atorvastatin in patients with hypercholesterolemia: A randomized clinical trial. Atherosclerosis 2021, 320, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Teramoto, T.; Daida, H.; Ikewaki, K.; Maeda, Y.; Nakagomi, M.; Shirakawa, M.; Kakikawa, T.; Numaguchi, H.; Johnson-Levonas, A.O.; et al. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib in Japanese patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2016, 249, 215–223. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hindi, N.N.; Alenbawi, J.; Nemer, G. Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia. J. Pers. Med. 2021, 11, 877. https://doi.org/10.3390/jpm11090877
Hindi NN, Alenbawi J, Nemer G. Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia. Journal of Personalized Medicine. 2021; 11(9):877. https://doi.org/10.3390/jpm11090877
Chicago/Turabian StyleHindi, Nagham N., Jamil Alenbawi, and Georges Nemer. 2021. "Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia" Journal of Personalized Medicine 11, no. 9: 877. https://doi.org/10.3390/jpm11090877
APA StyleHindi, N. N., Alenbawi, J., & Nemer, G. (2021). Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia. Journal of Personalized Medicine, 11(9), 877. https://doi.org/10.3390/jpm11090877