
Parsing Fabry Disease Metabolic Plasticity Using Metabolomics

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Blood Samples
2.2. Targeted Metabolomics Analysis
2.3. Plasma LysoGb3 Analysis
2.4. Alpha-D-Galactopyranosidase Activity Analysis
2.5. Data Analysis
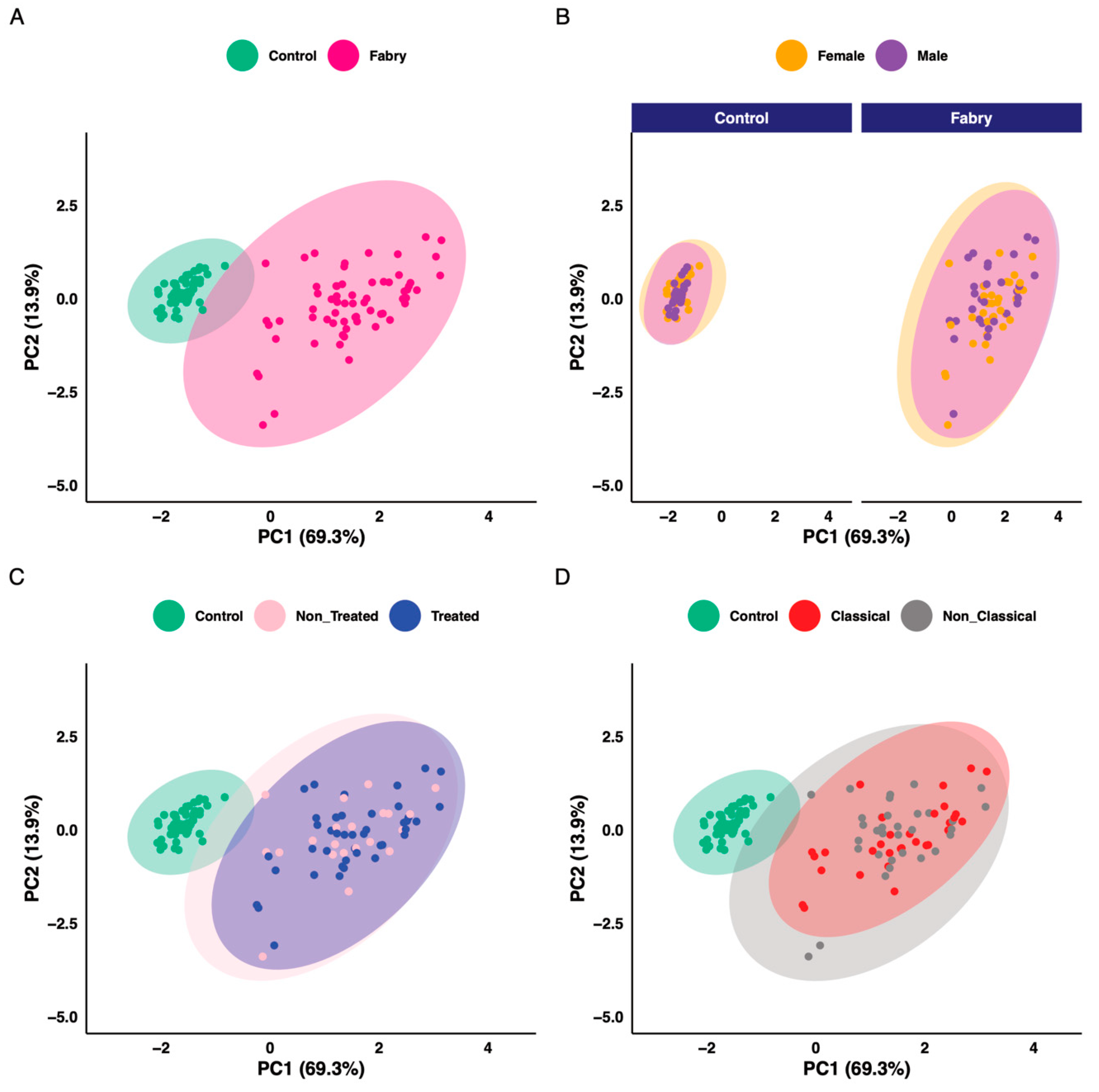
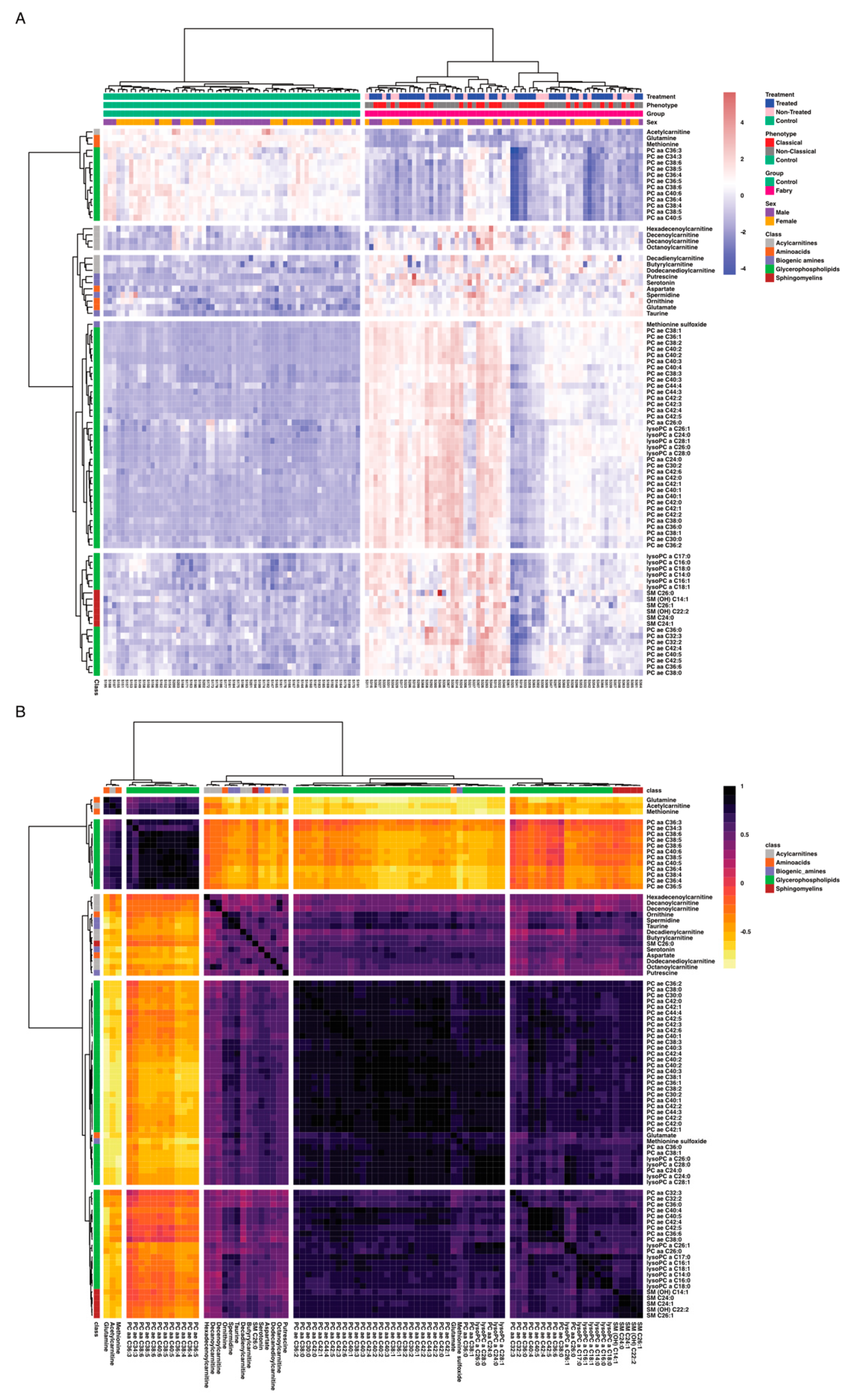
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of classical and nonclassical fabry disease: A multicenter study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hattori, K.; Ihara, K.; Ishii, A.; Nakamura, K.; Hirose, S. Newborn screening for fabry disease in Japan: Prevalence and genotypes of fabry disease in a pilot study. J. Hum. Genet. 2013, 58, 548. [Google Scholar] [CrossRef] [Green Version]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of fabry disease revealed by newborn screening in the taiwan chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.; Weidemann, F. Biomarkers for diagnosing and staging of fabry disease. Curr. Med. Chem. 2018, 25, 1530–1537. [Google Scholar] [CrossRef]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with fabry disease: Findings from the fabry registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.S.; Mehta, A.B. Difficulties and barriers in diagnosing fabry disease: What can be learnt from the literature? Expert Opin. Med. Diagn. 2013, 7, 589–599. [Google Scholar] [CrossRef]
- Zar-Kessler, C.; Karaa, A.; Sims, K.B.; Clarke, V.; Kuo, B. Understanding the gastrointestinal manifestations of fabry disease: Promoting prompt diagnosis. Ther. Adv. Gastroenterol. 2016, 9, 626–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winchester, B.; Young, E. Biochemical and Genetic Diagnosis of Fabry Disease; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Desnick, R.J.; Allen, K.Y.; Desnick, S.J.; Raman, M.K.; Bernlohr, R.W.; Krivit, W. Fabry’s disease: Enzymatic diagnosis of hemizygotes and heterozygotes: A-galactosidase activities in plasma, serum, urine, and leukocytes. J. Lab. Clin. Med. 1973, 81, 157–171. [Google Scholar] [PubMed]
- Chamoles, N.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 1, 195–196. [Google Scholar] [CrossRef]
- Desnick, R.J. A-galactosidase a deficiency: Fabry disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Sly, D., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 3, pp. 3733–3774. [Google Scholar]
- Mills, K.; Johnson, A.; Winchester, B. Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in fabry disease by tandem mass spectrometry. FEBS Lett. 2002, 515, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.; Mechtler, T.; Kasper, D.C.; Desnick, R.J. Correlation of lyso-gb3 levels in dried blood spots and sera from patients with classic and later-onset fabry disease. Mol. Genet. Metab. 2017, 121, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Boscaro, F.; Pieraccini, G.; Marca, G.l.; Bartolucci, G.; Luceri, C.; Luceri, F.; Moneti, G. Rapid quantitation of globotriaosylceramide in human plasma and urine: A potential application for monitoring enzyme replacement therapy in anderson-fabry disease. Rapid Commun. Mass Spectrom. 2002, 16, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E. Elevated globotriaosylsphingosine is a hallmark of fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, A.; Nicholls, K.; Fletcher, J.M.; Fuller, M. A simple method for quantification of plasma globotriaosylsphingosine: Utility for fabry disease. Mol. Genet. Metab. 2017, 122, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Aerts, J.M.; Auray-Blais, C.; Maruyama, H.; Mehta, A.B.; Skuban, N.; Krusinska, E.; Schiffmann, R. Assessment of plasma lyso-gb3 for clinical monitoring of treatment response in migalastat-treated patients with fabry disease. Genet. Med. 2021, 23, 192–201. [Google Scholar] [CrossRef]
- Mehta, A. Fabry disease: A review of current enzyme replacement strategies. Expert Opin. Orphan Drugs 2015, 3, 1319–1330. [Google Scholar] [CrossRef]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Felis, A.; Whitlow, M.; Kraus, A.; Warnock, D.G.; Wallace, E. Current and investigational therapeutics for fabry disease. Kidney Int. Rep. 2020, 5, 407–413. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Migalastat: A review in fabry disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arends, M.; Hollak, C.E.M.; Biegstraaten, M. Quality of life in patients with fabry disease: A systematic review of the literature. Orphanet J. Rare Dis. 2015, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Arends, M.; Biegstraaten, M.; Hughes, D.A.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Vaz, F.M.; van Kuilenburg, A.B.P.; Wanner, C.; et al. Retrospective study of long-term outcomes of enzyme replacement therapy in fabry disease: Analysis of prognostic factors. PLoS ONE 2017, 12, e0182379. [Google Scholar] [CrossRef]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. The large phenotypic spectrum of fabry disease requires graduated diagnosis and personalized therapy: A meta-analysis can help to differentiate missense mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef] [Green Version]
- Sudrie-Arnaud, B.; Marguet, F.; Patrier, S.; Martinovic, J.; Louillet, F.; Broux, F.; Charbonnier, F.; Dranguet, H.; Coutant, S.; Vezain, M.; et al. Metabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 481, 1–8. [Google Scholar] [CrossRef]
- Tebani, A.; Schmitz-Afonso, I.; Abily-Donval, L.; Heron, B.; Piraud, M.; Ausseil, J.; Brassier, A.; De Lonlay, P.; Zerimech, F.; Vaz, F.M.; et al. Urinary metabolic phenotyping of mucopolysaccharidosis type i combining untargeted and targeted strategies with data modeling. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 475, 7–14. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-based strategies in precision medicine: Toward a paradigm shift in inborn errors of metabolism investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [Green Version]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical metabolomics: The new metabolic window for inborn errors of metabolism investigations in the post-genomic era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in pbmc isolated from patients with fabry disease: Preliminary findings. Mol. BioSyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated inflammatory plasma biomarkers in patients with fabry disease: A critical link to heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [Green Version]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early markers of fabry disease revealed by proteomics. Mol. BioSyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef]
- Manwaring, V.; Heywood, W.E.; Clayton, R.; Lachmann, R.H.; Keutzer, J.; Hindmarsh, P.; Winchester, B.; Heales, S.; Mills, K. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: Evidence of presymptomatic kidney disease in pediatric fabry and type-i diabetic patients. J. Proteome Res. 2013, 12, 2013–2021. [Google Scholar] [CrossRef]
- Heo, S.H.; Kang, E.; Kim, Y.M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.H.; Kim, J.M.; Choi, I.H.; et al. Fabry disease: Characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [Green Version]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A human stem cell model of fabry disease implicates limp-2 accumulation in cardiomyocyte pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; et al. Generation of gla-knockout human embryonic stem cell lines to model autophagic dysfunction and exosome secretion in fabry disease-associated hypertrophic cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Beer, M.; Kralewski, M.; Siwy, J.; Kampmann, C. Early detection of organ involvement in fabry disease by biomarker assessment in conjunction with lge cardiac mri: Results from the sophia study. Mol. Genet. Metab. 2019, 126, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Doykov, I.D.; Heywood, W.E.; Nikolaenko, V.; Śpiewak, J.; Hällqvist, J.; Clayton, P.T.; Mills, P.; Warnock, D.G.; Nowak, A.; Mills, K. Rapid, proteomic urine assay for monitoring progressive organ disease in fabry disease. J. Med. Genet. 2020, 57, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Mauhin, W.; Abily-Donval, L.; Lesueur, C.; Berger, M.G.; Nadjar, Y.; Berger, J.; Benveniste, O.; Lamari, F.; Laforêt, P.; et al. A proteomics-based analysis reveals predictive biological patterns in fabry disease. J. Clin. Med. 2020, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A metabolomic study to identify new globotriaosylceramide-related biomarkers in the plasma of fabry disease patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef]
- Mauhin, W.; Lidove, O.; Amelin, D.; Lamari, F.; Caillaud, C.; Mingozzi, F.; Dzangue-Tchoupou, G.; Arouche-Delaperche, L.; Douillard, C.; Dussol, B.; et al. Deep characterization of the anti-drug antibodies developed in fabry disease patients, a prospective analysis from the french multicenter cohort ffabry. Orphanet J. Rare Dis. 2018, 13, 127. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, S.L.; Guggenbichler, W.; Weinberger, K.M.; Graber, A.; Stoeggl, W.M. Device for Quantitative Analysis of a Drug or Metabolite Profile. Google Patents WO2007003344A2, 11 January 2007. [Google Scholar]
- Ramsay, S.L.; Stoeggl, W.M.; Weinberger, K.M.; Graber, A.; Guggenbichler, W. Apparatus and Method for Analyzing a Metabolite Profile. Google Patents US8265877B2, 11 September 2012. [Google Scholar]
- Van Den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; Van Der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Liang, F.; Song, Q.; Qiu, P. An equivalent measure of partial correlation coefficients for high-dimensional gaussian graphical models. J. Am. Stat. Assoc. 2015, 110, 1248–1265. [Google Scholar] [CrossRef]
- Thistlethwaite, L.R.; Petrosyan, V.; Li, X.; Miller, M.J.; Elsea, S.H.; Milosavljevic, A. Ctd: An information-theoretic algorithm to interpret sets of metabolomic and transcriptomic perturbations in the context of graphical models. PLoS Comput. Biol. 2021, 17, e1008550. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.N.; Ziegler, A. Ranger: A fast implementation of random forests for high dimensional data in C++ and r. J. Stat. Softw. 2017, 77, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Caret: Classification and Regression Training; Astrophysics Source Code Library: 2020. Avaliable online: https://ascl.net/ (accessed on 1 May 2021).
- Eriksson, L.; Trygg, J.; Wold, S. A chemometrics toolbox based on projections and latent variables. J. Chemom. 2014, 28, 332–346. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, F.; Wang, Z.; Lu, Y.; Xu, Y.; Wang, K.; Shen, H.; Yang, P.; Li, S.; et al. Quantitative profiling of glycerophospholipids during mouse and human macrophage differentiation using targeted mass spectrometry. Sci. Rep. 2017, 7, 412. [Google Scholar] [CrossRef] [Green Version]
- Kosicek, M.; Hecimovic, S. Phospholipids and Alzheimer’s disease: Alterations, mechanisms and potential biomarkers. Int. J. Mol. Sci. 2013, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Miletic Vukajlovic, J.; Drakulic, D.; Pejic, S.; Ilic, T.V.; Stefanovic, A.; Petkovic, M.; Schiller, J. Increased plasma phosphatidylcholine/lysophosphatidylcholine ratios in patients with Parkinson’s disease. Rapid Commun. Mass Spectrom. 2019, 34, e8595. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Hazen, S.L. Lipid oxidation and cardiovascular disease: Introduction to a review series. Circ. Res. 2010, 107, 1167–1169. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Investig. 1991, 88, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.; Sillence, D.O.; Tchan, M.C. Homocysteine and erythrocyte sedimentation rate correlate with cerebrovascular disease in fabry disease. JIMD Rep. 2012, 6, 101–105. [Google Scholar] [PubMed] [Green Version]
- Demuth, K.; Germain, D.P. Endothelial markers and homocysteine in patients with classic fabry disease. Acta Paediatr. Suppl. 2002, 91, 57–61. [Google Scholar] [CrossRef]
- Fedi, S.; Gensini, F.; Gori, A.M.; Abbate, R.; Borsini, W. Homocysteine and tissue factor pathway inhibitor levels in patients with fabry’s disease. J. Thromb. Haemost. 2005, 3, 2117–2119. [Google Scholar] [CrossRef] [Green Version]
- Van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 2006, 21, 1161–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaoui, M.; Boutin, M.; Lavoie, P.; Auray-Blais, C. Tandem mass spectrometry multiplex analysis of methylated and non-methylated urinary gb3 isoforms in fabry disease patients. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 452, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Sidransky, E.; Tayebi, N. The role of epigenetics in lysosomal storage disorders: Uncharted territory. Mol. Genet. Metab. 2017, 122, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerrière, A.L.; Barbey, F. Autophagosome maturation is impaired in fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, H.; Hossain, M.A.; Miyajima, T.; Nagao, K.; Miyashita, T.; Eto, Y. Dysregulated DNA methylation of gla gene was associated with dysfunction of autophagy. Mol. Genet. Metab. 2019, 126, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Jacques, C.E.; Hammerschmidt, T.; de Souza, H.M.; Donida, B.; Deon, M.; Vairo, F.P.; Lourenco, C.M.; Giugliani, R.; Vargas, C.R. Biomolecules damage and redox status abnormalities in fabry patients before and during enzyme replacement therapy. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 461, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Moras, A.M.; Reinhardt, L.S.; Busatto, F.F.; de Moura Sperotto, N.D.; Saffi, J.; Moura, D.J.; Giugliani, R.; Vargas, C.R. Globotriaosylsphingosine induces oxidative DNA damage in cultured kidney cells. Nephrology 2017, 22, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.B.; Galdieri, L.C.; Pereira, V.G.; Martins, A.M.; D’Almeida, V. Evaluation of oxidative stress markers and cardiovascular risk factors in fabry disease patients. Genet. Mol. Biol. 2012, 35, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calo, L.A. Oxidative stress and the altered reaction to it in fabry disease: A possible target for cardiovascular-renal remodeling? PLoS ONE 2018, 13, e0204618. [Google Scholar]
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calo, L.A. Oxidative stress and cardiovascular-renal damage in fabry disease: Is there room for a pathophysiological involvement? J. Clin. Med. 2018, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Loso, J.; Lund, N.; Avanesov, M.; Muschol, N.; Lezius, S.; Cordts, K.; Schwedhelm, E.; Patten, M. Serum biomarkers of endothelial dysfunction in fabry associated cardiomyopathy. Front. Cardiovasc. Med. 2018, 5, 108. [Google Scholar] [CrossRef]
- Satoh, K. Globotriaosylceramide induces endothelial dysfunction in fabry disease. Arter. Thromb. Vasc. Biol. 2014, 34, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Luscher, T.F.; Camici, G.G. Globotriaosylsphingosine accumulation and not alpha-galactosidase—A deficiency causes endothelial dysfunction in fabry disease. PLoS ONE 2012, 7, e36373. [Google Scholar] [CrossRef]
- Shen, J.S.; Arning, E.; West, M.L.; Day, T.S.; Chen, S.; Meng, X.L.; Forni, S.; McNeill, N.; Goker-Alpan, O.; Wang, X.; et al. Tetrahydrobiopterin deficiency in the pathogenesis of fabry disease. Hum. Mol. Genet. 2017, 26, 1182–1192. [Google Scholar] [CrossRef]
- Tseng, W.L.; Chou, S.J.; Chiang, H.C.; Wang, M.L.; Chien, C.S.; Chen, K.H.; Leu, H.B.; Wang, C.Y.; Chang, Y.L.; Liu, Y.Y.; et al. Imbalanced production of reactive oxygen species and mitochondrial antioxidant sod2 in fabry disease-specific human induced pluripotent stem cell-differentiated vascular endothelial cells. Cell Transpl. 2017, 26, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Oien, D.B.; Moskovitz, J. Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr. Top. Dev. Biol. 2008, 80, 93–133. [Google Scholar]
- Stadtman, E.R.; Van Remmen, H.; Richardson, A.; Wehr, N.B.; Levine, R.L. Methionine oxidation and aging. Biochim. Biophys. Acta 2005, 1703, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kodera, Y.; Saito, T.; Fujimoto, K.; Momozono, A.; Hayashi, A.; Kamata, Y.; Shichiri, M. Methionine sulfoxides in serum proteins as potential clinical biomarkers of oxidative stress. Sci. Rep. 2016, 6, 38299. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ducatez, F.; Mauhin, W.; Boullier, A.; Pilon, C.; Pereira, T.; Aubert, R.; Benveniste, O.; Marret, S.; Lidove, O.; Bekri, S.; et al. Parsing Fabry Disease Metabolic Plasticity Using Metabolomics. J. Pers. Med. 2021, 11, 898. https://doi.org/10.3390/jpm11090898
Ducatez F, Mauhin W, Boullier A, Pilon C, Pereira T, Aubert R, Benveniste O, Marret S, Lidove O, Bekri S, et al. Parsing Fabry Disease Metabolic Plasticity Using Metabolomics. Journal of Personalized Medicine. 2021; 11(9):898. https://doi.org/10.3390/jpm11090898
Chicago/Turabian StyleDucatez, Franklin, Wladimir Mauhin, Agnès Boullier, Carine Pilon, Tony Pereira, Raphaël Aubert, Olivier Benveniste, Stéphane Marret, Olivier Lidove, Soumeya Bekri, and et al. 2021. "Parsing Fabry Disease Metabolic Plasticity Using Metabolomics" Journal of Personalized Medicine 11, no. 9: 898. https://doi.org/10.3390/jpm11090898
APA StyleDucatez, F., Mauhin, W., Boullier, A., Pilon, C., Pereira, T., Aubert, R., Benveniste, O., Marret, S., Lidove, O., Bekri, S., & Tebani, A. (2021). Parsing Fabry Disease Metabolic Plasticity Using Metabolomics. Journal of Personalized Medicine, 11(9), 898. https://doi.org/10.3390/jpm11090898