
Which Therapy for Non-Type(T)2/T2-Low Asthma


{kind=link}
{kind=link}
Abstract
:1. Introduction
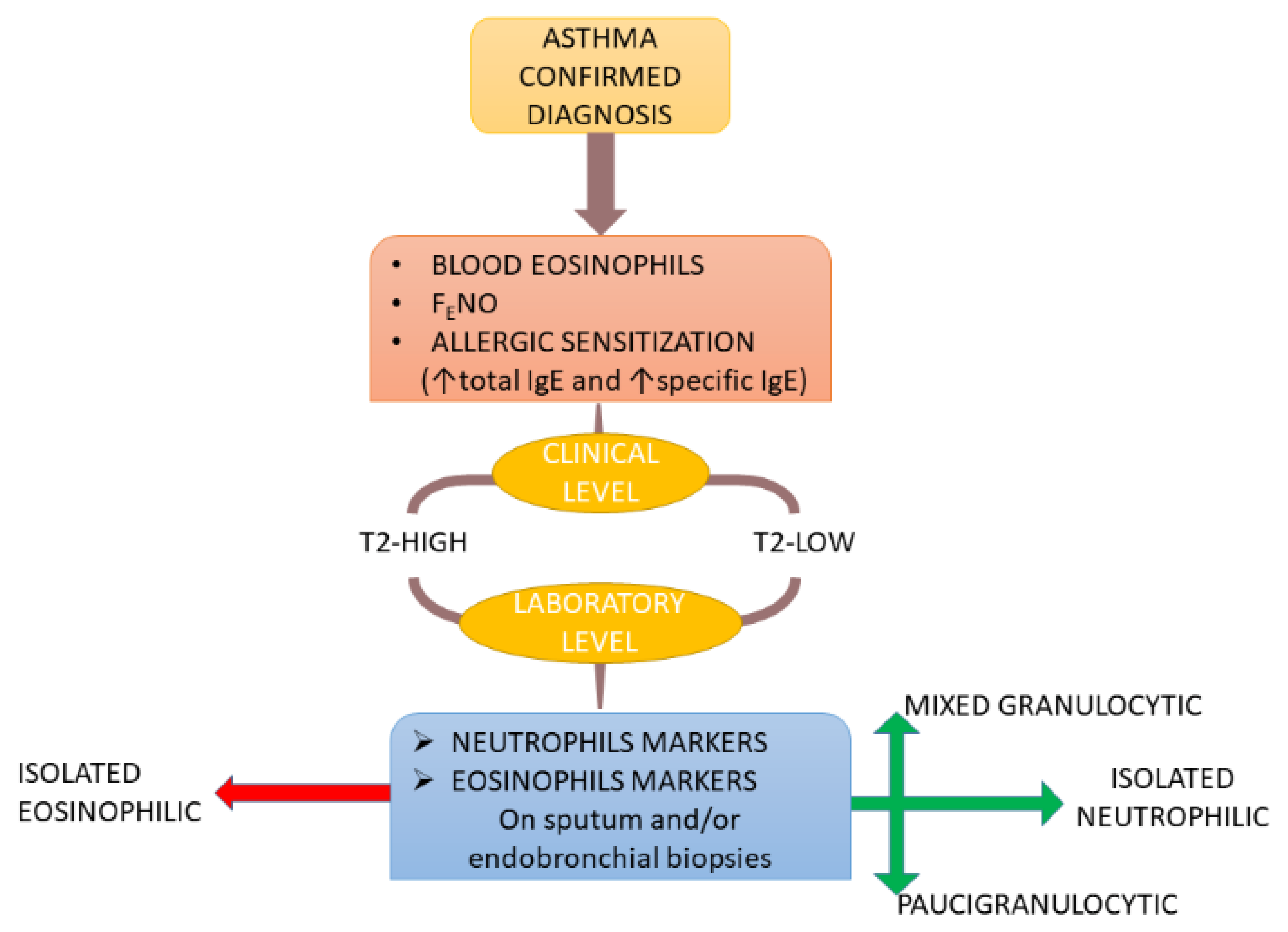
2. Stratification of T2-Low Asthma Patients
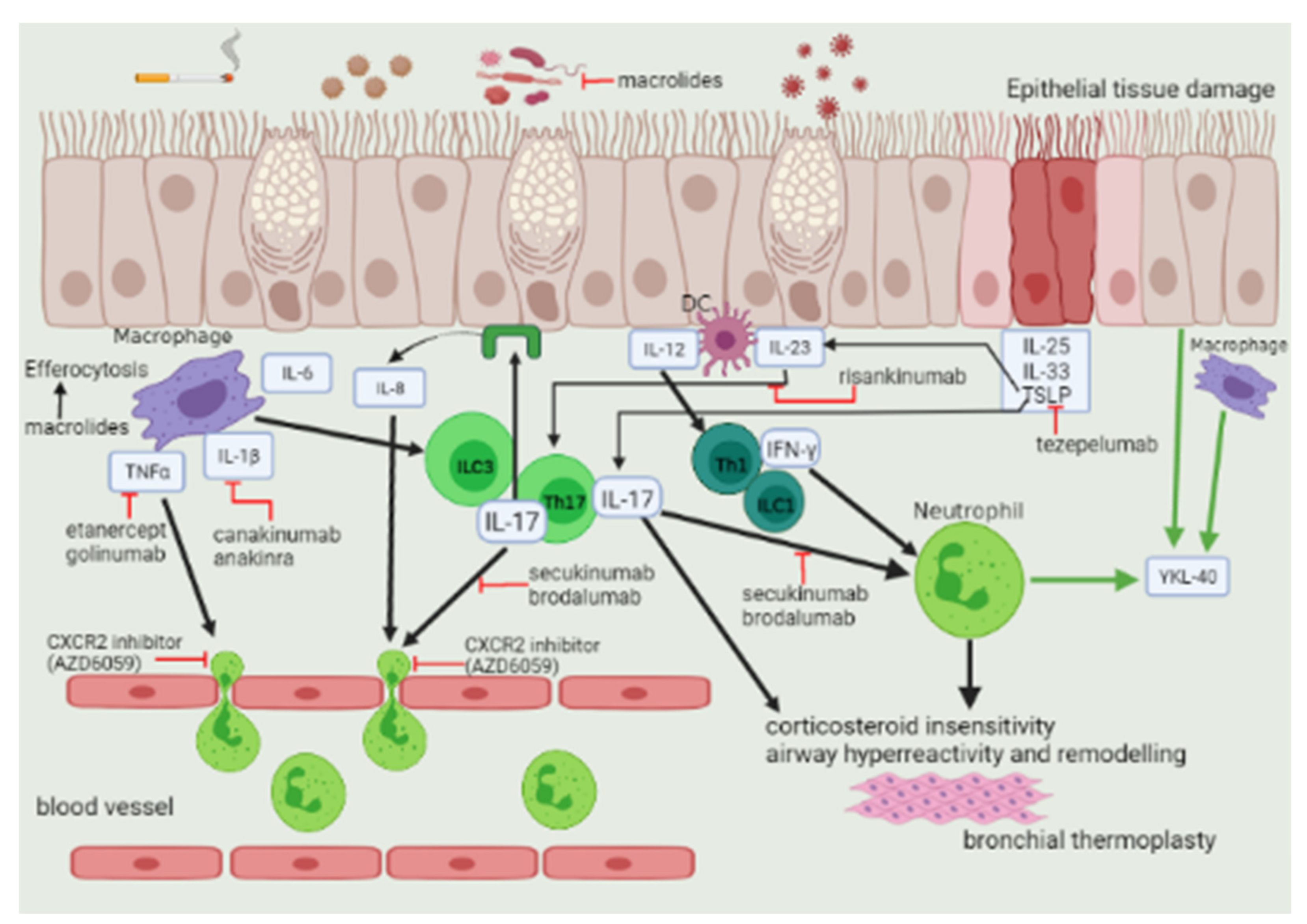
3. Mechanisms of T2-Low Asthma
3.1. Type 1 Immunity
3.2. Type 3 Immunity
4. IL-6 and Obesity
5. Paucigranulocytic Asthma
6. Non-T2 Biomarkers
7. Therapeutic Strategy
7.1. Non-Pharmacological Intervention
7.2. Bronchial Thermoplasty
7.3. Macrolides
7.4. Long-Acting Muscarinic Antagonists (LAMA)
7.5. Biological Agents
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tliba, O.; Panettieri, R.A., Jr. Paucigranulocytic asthma: Uncoupling of airway obstruction from inflammation. J. Allergy Clin. Immunol. 2019, 143, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druilhe, A.; Létuvé, S.; Pretolani, M. Glucocorticoid-induced apoptosis in human eosinophils: Mechanisms of action. Apoptosis 2003, 8, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Ashdown, H.; Gounni, A.S. The Molecular Mechanisms of Glucocorticoids-Mediated Neutrophil Survival. Curr. Drug Targets 2011, 12, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Cowan, D.C.; Cowan, J.O.; Palmay, R.; Williamson, A.; Taylor, D.R. Effects of steroid therapy on inflammatory cell subtypes in asthma. Thorax 2010, 65, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Ricciardolo, F.L.M.; Sprio, A.E.; Baroso, A.; Gallo, F.; Riccardi, E.; Bertolini, F.; Carriero, V.; Arrigo, E.; Ciprandi, G. Characterization of T2-Low and T2-High Asthma Phenotypes in Real-Life. Biomedicines 2021, 9, 1684. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Heaney, L.G.; Busby, J.; Hanratty, C.E.; Djukanovic, R.; Woodcock, A.; Walker, S.M.; Hardman, T.; Arron, J.R.; Choy, D.F.; Bradding, P.; et al. Composite type-2 biomarker strategy versus a symptom–risk-based algorithm to adjust corticosteroid dose in patients with severe asthma: A multicentre, single-blind, parallel group, randomised controlled trial. Lancet Respir. Med. 2021, 9, 57–68. [Google Scholar] [CrossRef]
- Carr, T.F. Treatment approaches for the patient with T2 low Asthma. Ann. Allergy Asthma Immunol. 2021, 127, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Hinks, T.S.; Levine, S.J.; Brusselle, G.G. Treatment options in type-2 low asthma. Eur. Respir. J. 2021, 57, 2000528. [Google Scholar] [CrossRef] [PubMed]
- Schleich, F.N.; Manise, M.; Sele, J.; Henket, M.; Seidel, L.; Louis, R. Distribution of sputum cellular phenotype in a large asthma cohort: Predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm. Med. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanevello, A.; Confalonieri, M.; Sulotto, F.; Romano, F.; Balzano, G.; Migliori, G.B.; Bianchi, A.; Michetti, G. Induced Sputum Cellularity. Am. J. Respir. Crit. Care Med. 2000, 162, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Scott, R.; Boyle, M.J.; Gibson, P.G. Inflammatory subtypes in asthma: Assessment and identification using induced sputum. Respirology 2006, 11, 54–61. [Google Scholar] [CrossRef]
- Kupczyk, M.; Dahlén, B.; Sterk, P.J.; Nizankowska-Mogilnicka, E.; Papi, A.; Bel, E.H.; Chanez, P.; Howarth, P.H.; Holgate, S.T.; Brusselle, G.; et al. Stability of phenotypes defined by physiological variables and biomarkers in adults with asthma. Allergy 2014, 69, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.R.; Van Dalen, C.J.; Harding, E.; Hermans, I.F.; Douwes, J. Effects of treatment changes on asthma phenotype prevalence and airway neutrophil function. BMC Pulm. Med. 2017, 17, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.E.; Schwartz, L.B.; Langmack, E.; Halliday, J.L.; Trudeau, J.B.; Gibbs, R.L.; Chu, H.W. Evidence That Severe Asthma Can Be Divided Pathologically into Two Inflammatory Subtypes with Distinct Physiologic and Clinical Characteristics. Am. J. Respir. Crit. Care Med. 1999, 160, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Sorbello, V.; Folino, A.; Gallo, F.; Massaglia, G.M.; Favatà, G.; Conticello, S.; Vallese, D.; Gani, F.; Malerba, M.; et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J. Allergy Clin. Immunol. 2017, 140, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullone, M.; Carriero, V.; Bertolini, F.; Folino, A.; Mannelli, A.; Di Stefano, A.; Gnemmi, I.; Torchio, R.; Ricciardolo, F.L. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur. Respir. J. 2019, 54, 1900068. [Google Scholar] [CrossRef]
- Green, R.H.; Brightling, C.; Woltmann, G.; Parker, D.; Wardlaw, A.; Pavord, I. Analysis of induced sputum in adults with asthma: Identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 2002, 57, 875–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.F.; Kraft, M. Use of biomarkers to identify phenotypes and endotypes of severe asthma. Ann. Allergy, Asthma Immunol. 2018, 121, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Volder, J.; Vereecke, L.; Joos, G.; Maes, T. Targeting neutrophils in asthma: A therapeutic opportunity? Biochem. Pharmacol. 2020, 182, 114292. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.; Nong, G.; Ward, J.; Seumois, G.; Prince, L.; Wilson, S.J.; Cornelius, V.; Dent, G.; Djukanovic, R. Prosurvival activity for airway neutrophils in severe asthma. Thorax 2010, 65, 684–689. [Google Scholar] [CrossRef] [Green Version]
- Berry, A.; Busse, W.W. Biomarkers in asthmatic patients: Has their time come to direct treatment? J. Allergy Clin. Immunol. 2016, 137, 1317–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciprandi, G.; Tosca, M.A.; Silvestri, M.; Ricciardolo, F.L.M. Inflammatory biomarkers in asthma endotypes and consequent personalized therapy. Expert Rev. Clin. Immunol. 2017, 13, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Fitzpatrick, A.M.; Chipps, B.E.; Holguin, F.; Woodruff, P.G. T2-“Low” Asthma: Overview and Management Strategies. J. Allergy Clin. Immunol. Pr. 2020, 8, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Sze, E.; Bhalla, A.; Nair, P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy 2020, 75, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britt, R.D.; Thompson, M.A.; Sasse, S.K.; Pabelick, C.M.; Gerber, A.N.; Prakash, Y.S. Th1 cytokines TNF-α and IFN-γ promote corticosteroid resistance in developing human airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L71–L81. [Google Scholar] [CrossRef]
- Guida, G.; Riccio, A.M. Immune induction of airway remodeling. Semin. Immunol. 2019, 46, 101346. [Google Scholar] [CrossRef] [PubMed]
- Hudey, S.N.; Ledford, D.K.; Cardet, J.C. Mechanisms of non-type 2 asthma. Curr. Opin. Immunol. 2020, 66, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Samitas, K.; Zervas, E.; Gaga, M. T2-low asthma. Curr. Opin. Pulm. Med. 2017, 23, 48–55. [Google Scholar] [CrossRef]
- Zijlstra, G.J.; Hacken, N.H.T.T.; Hoffmann, R.F.; van Oosterhout, A.J.M.; Heijink, I.H. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur. Respir. J. 2012, 39, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Tello, A.; Halwani, R.; Hamid, Q.; Al-Muhsen, S. Glucocorticoid Receptor-Beta Up-Regulation and Steroid Resistance Induction by IL-17 and IL-23 Cytokine Stimulation in Peripheral Mononuclear Cells. J. Clin. Immunol. 2012, 33, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Irvin, C.; Zafar, I.; Good, J.; Rollins, D.; Christianson, C.; Gorska, M.M.; Martin, R.J.; Alam, R. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J. Allergy Clin. Immunol. 2014, 134, 1175–1186.e7. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Voo, K.S.; Liu, B.; Chen, C.-Y.; Uygungil, B.; Spoede, W.; Bernstein, J.A.; Huston, D.P.; Liu, Y.-J. A novel subset of CD4+ TH2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J. Exp. Med. 2010, 207, 2479–2491. [Google Scholar] [CrossRef]
- Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Annunziato, F. Th17 plasticity: Pathophysiology and treatment of chronic inflammatory disorders. Curr. Opin. Pharmacol. 2014, 17, 12–16. [Google Scholar] [CrossRef]
- Choy, D.F.; Hart, K.M.; Borthwick, L.A.; Shikotra, A.; Nagarkar, D.R.; Siddiqui, S.; Jia, G.; Ohri, C.M.; Doran, E.; Vannella, K.M.; et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 2015, 7, 301ra129. [Google Scholar] [CrossRef] [Green Version]
- Detoraki, A.; Granata, F.; Staibano, S.; Rossi, F.W.; Marone, G.; Genovese, A. Angiogenesis and lymphangiogenesis in bronchial asthma. Allergy 2010, 65, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, H.; Gao, R.; Gao, X.; Xu, F.; Wang, Q.; Lu, G.; Xia, D.; Zhou, J. IL-17A, But Not IL-17F, Is Indispensable for Airway Vascular Remodeling Induced by Exaggerated Th17 Cell Responses in Prolonged Ovalbumin-Challenged Mice. J. Immunol. 2015, 194, 3557–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postma, D.S.; Timens, W. Remodeling in Asthma and Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2006, 3, 434–439. [Google Scholar] [CrossRef]
- Ribatti, D.; Puxeddu, I.; Crivellato, E.; Nico, B.; Vacca, A.; Levi-Schaffer, F. Angiogenesis in asthma. Clin. Exp. Allergy 2009, 39, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Samitas, K.; Poulos, N.; Semitekolou, M.; Morianos, I.; Tousa, S.; Economidou, E.; Robinson, D.S.; Kariyawasam, H.H.; Zervas, E.; Corrigan, C.; et al. Activin-A is overexpressed in severe asthma and is implicated in angiogenic processes. Eur. Respir. J. 2016, 47, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Ricciardolo, F.L.; Folkerts, G.; Folino, A.; Mognetti, B. Bradykinin in asthma: Modulation of airway inflammation and remodelling. Eur. J. Pharmacol. 2018, 827, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M.; Sabatini, F.; Sorbello, V.; Benedetto, S.; Defilippi, I.; Petecchia, L.; Usai, C.; Gnemmi, I.; Balbi, B.; De Rose, V.; et al. Expression of vascular remodelling markers in relation to bradykinin receptors in asthma and COPD. Thorax 2013, 68, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Alwan, A.; Bates, J.H.T.; Chapman, D.G.; Kaminsky, D.A.; DeSarno, M.J.; Irvin, C.G.; Dixon, A.E. The Nonallergic Asthma of Obesity. A Matter of Distal Lung Compliance. Am. J. Respir. Crit. Care Med. 2014, 189, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, H.A.; Gibson, P.G.; Garg, M.L.; Wood, L.G. Airway inflammation is augmented by obesity and fatty acids in asthma. Eur. Respir. J. 2011, 38, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Sideleva, O.; Suratt, B.T.; Black, K.E.; Tharp, W.G.; Pratley, R.E.; Forgione, P.; Dienz, O.; Irvin, C.G.; Dixon, A.E. Obesity and Asthma. Am. J. Respir. Crit. Care Med. 2012, 186, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.F.; Zeki, A.A.; Kraft, M. Eosinophilic and Noneosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 22–37. [Google Scholar] [CrossRef]
- Jonckheere, A.-C.; Bullens, D.M.A.; Seys, S.F. Innate lymphoid cells in asthma: Pathophysiological insights from murine models to human asthma phenotypes. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 53–60. [Google Scholar] [CrossRef]
- Peters, M.C.; McGrath, K.W.; Hawkins, G.A.; Hastie, A.T.; Levy, B.D.; Israel, E.; Phillips, B.R.; Mauger, D.T.; Comhair, S.A.; Erzurum, S.C.; et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: A cross-sectional analysis of two cohorts. Lancet Respir. Med. 2016, 4, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Turan, N.; Edwards, M.J.; Bates, S.; Shaw, D.; Chung, K.F.; Loza, M.J.; James, A.; Van Oosterhout, A. the U-BIOPRED Study Group IL-6 pathway upregulation in subgroup of severe asthma is associated with neutrophilia and poor lung function. Clin. Exp. Allergy 2018, 48, 475–478. [Google Scholar] [CrossRef]
- Cauvi, D.M.; Cauvi, G.; Toomey, C.B.; Jacquinet, E.; Pollard, K.M. From the Cover: Interplay Between IFN-γ and IL-6 Impacts the Inflammatory Response and Expression of Interferon-Regulated Genes in Environmental-Induced Autoimmunity. Toxicol. Sci. 2017, 158, 227–239. [Google Scholar] [CrossRef]
- McLoughlin, R.; Witowski, J.; Robson, R.L.; Wilkinson, T.; Hurst, S.M.; Williams, A.S.; Williams, J.D.; Rose-John, S.; Jones, S.A.; Topley, N. Interplay between IFN-γ and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J. Clin. Investig. 2003, 112, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; He, X.Y.; Baines, K.; Gunawardhana, L.P.; Simpson, J.L.; Li, F.; Gibson, P.G. Different inflammatory phenotypes in adults and children with acute asthma. Eur. Respir. J. 2011, 38, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntontsi, P.; Loukides, S.; Bakakos, P.; Kostikas, K.; Papatheodorou, G.; Papathanassiou, E.; Hillas, G.; Koulouris, N.; Papiris, S.; Papaioannou, A.I. Clinical, functional and inflammatory characteristics in patients with paucigranulocytic stable asthma: Comparison with different sputum phenotypes. Allergy 2017, 72, 1761–1767. [Google Scholar] [CrossRef]
- Demarche, S.; Schleich, F.; Henket, M.; Paulus, V.; Van Hees, T.; Louis, R. Detailed analysis of sputum and systemic inflammation in asthma phenotypes: Are paucigranulocytic asthmatics really non-inflammatory? BMC Pulm. Med. 2016, 16, 46. [Google Scholar] [CrossRef] [Green Version]
- Slats, A.M.; Janssen, K.; Van Schadewijk, A.; Van Der Plas, D.T.; Schot, R.; Aardweg, J.G.V.D.; De Jongste, J.C.; Hiemstra, P.; Mauad, T.; Rabe, K.F.; et al. Bronchial Inflammation and Airway Responses to Deep Inspiration in Asthma and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 176, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P.; Walls, A.; Holgate, S.T. The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 2006, 117, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, I.; Louis, R.; Manise, M.; Dentener, M.A.; Irvin, C.G.; Janssen-Heininger, Y.M.; Brightling, C.E.; Wouters, E.F.; Reynaert, N.L. Increased glutaredoxin-1 and decreased protein S-glutathionylation in sputum of asthmatics. Eur. Respir. J. 2013, 41, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agache, I.; Eguiluz-Gracia, I.; Cojanu, C.; Laculiceanu, A.; del Giacco, S.; Zemelka-Wiacek, M.; Kosowska, A.; Akdis, C.A.; Jutel, M. Advances and highlights in asthma in 2021. Allergy 2021, 76, 3390–3407. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; Maestrelli, P.; Turato, G.; Mapp, C.E.; Milani, G.; Pivirotto, F.; Fabbri, L.M.; Di Stefano, A. Airway wall remodeling after cessation of exposure to isocyanates in sensitized asthmatic subjects. Am. J. Respir. Crit. Care Med. 1995, 151, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Medrek, S.; Parulekar, A.D.; Hanania, N.A. Predictive Biomarkers for Asthma Therapy. Curr. Allergy Asthma Rep. 2017, 17, 17–69. [Google Scholar] [CrossRef] [PubMed]
- Nadif, R.; Siroux, V.; Boudier, A.; le Moual, N.; Just, J.; Gormand, F.; Pison, C.; Matran, R.; Pin, I. Blood granulocyte patterns as predictors of asthma phenotypes in adults from the EGEA study. Eur. Respir. J. 2016, 48, 1040–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriero, V.; Bertolini, F.; Sprio, A.E.; Bullone, M.; Ciprandi, G.; Ricciardolo, F.L.M. High levels of plasma fibrinogen could predict frequent asthma exacerbations. J. Allergy Clin. Immunol. Pract. 2020, 8, 2392–2395.e7. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Strasser, D.S.; Pierlot, G.M.; Farine, H.; Izuhara, K.; Akdis, C.A. Monitoring inflammatory heterogeneity with multiple biomarkers for multidimensional endotyping of asthma. J. Allergy Clin. Immunol. 2018, 141, 442–445. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Humbert, M.; Buhl, R.; Cruz, A.A.; Inoue, H.; Korom, S.; Hanania, N.A.; Nair, P. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clin. Exp. Allergy 2017, 47, 161–175. [Google Scholar] [CrossRef]
- Hinks, T.S.; Brown, T.; Lau, L.C.; Rupani, H.; Barber, C.; Elliott, S.; Ward, J.A.; Ono, J.; Ohta, S.; Izuhara, K.; et al. Multidimensional endotyping in patients with severe asthma reveals inflammatory heterogeneity in matrix metalloproteinases and chitinase 3–like protein 1. J. Allergy Clin. Immunol. 2016, 138, 61–75. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.W.; Icitovic, N.; Boushey, H.A.; Lazarus, S.C.; Sutherland, E.R.; Chinchilli, V.M.; Fahy, J.V. A Large Subgroup of Mild-to-Moderate Asthma Is Persistently Noneosinophilic. Am. J. Respir. Crit. Care Med. 2012, 185, 612–619. [Google Scholar] [CrossRef]
- Maes, T.; Cobos, F.A.; Schleich, F.; Sorbello, V.; Henket, M.; De Preter, K.; Bracke, K.; Conickx, G.; Mesnil, C.; Vandesompele, J.; et al. Asthma inflammatory phenotypes show differential microRNA expression in sputum. J. Allergy Clin. Immunol. 2016, 137, 1433–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, J.L.; Chen, A.; Diaz, M.P.; Zirn, N.; Gupta, A.; Britto, C.; Sauler, M.; Yan, X.; Stewart, E.; Santerian, K.; et al. A Network of Sputum MicroRNAs Is Associated with Neutrophilic Airway Inflammation in Asthma. Am. J. Respir. Crit. Care Med. 2020, 202, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Schleich, F.; Demarche, S.; Louis, R. Biomarkers in the Management of Difficult Asthma. Curr. Top. Med. Chem. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.Y.; Pinkerton, J.W.; Gibson, P.; Cooper, M.; Horvat, J.C.; Hansbro, P. Inflammasomes in COPD and neutrophilic asthma. Thorax 2015, 70, 1199–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Sorbello, V.; Ciprandi, G.; Di Stefano, A.; Massaglia, G.M.; Favatà, G.; Conticello, S.; Malerba, M.; Folkerts, G.; Profita, M.; Rolla, G.; et al. Nasal IL-17F is related to bronchial IL-17F/neutrophilia and exacerbations in stable atopic severe asthma. Allergy 2015, 70, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.C. Novel approaches to the management of noneosinophilic asthma. Ther. Adv. Respir. Dis. 2016, 10, 211–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, O.; Le Moual, N. Do chronic workplace irritant exposures cause asthma? Curr. Opin. Allergy Clin. Immunol. 2016, 16, 75–85. [Google Scholar] [CrossRef]
- Maghni, K.; Lemière, C.; Ghezzo, H.; Yuquan, W.; Malo, J.-L. Airway Inflammation after Cessation of Exposure to Agents Causing Occupational Asthma. Am. J. Respir. Crit. Care Med. 2004, 169, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siew, L.Q.C.; Wu, S.-Y.; Ying, S.; Corrigan, C. Cigarette smoking increases bronchial mucosal IL-17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clin. Exp. Allergy 2017, 47, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, R.; Livingston, E.; McMahon, A.D.; Lafferty, J.; Fraser, I.; Spears, M.; McSharry, C.P.; Thomson, N. Effects of Smoking Cessation on Lung Function and Airway Inflammation in Smokers with Asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Baines, K.J.; Gibson, P.G.; Wood, L.G. Changes in Expression of Genes Regulating Airway Inflammation Following a High-Fat Mixed Meal in Asthmatics. Nutrients 2016, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.G.; Garg, M.L.; Gibson, P.G. A high-fat challenge increases airway inflammation and impairs bronchodilator recovery in asthma. J. Allergy Clin. Immunol. 2011, 127, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Boulet, L.-P.; Turcotte, H.; Martin, J.; Poirier, P. Effect of bariatric surgery on airway response and lung function in obese subjects with asthma. Respir. Med. 2012, 106, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Huisstede, A.; Rudolphus, A.; Cabezas, M.C.; Biter, L.U.; Van De Geijn, G.-J.; Taube, C.; Hiemstra, P.; Braunstahl, G.; Van Schadewijk, A. Effect of bariatric surgery on asthma control, lung function and bronchial and systemic inflammation in morbidly obese subjects with asthma. Thorax 2015, 70, 659–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretolani, M.; Bergqvist, A.; Thabut, G.; Dombret, M.-C.; Knapp, D.; Hamidi, F.; Alavoine, L.; Taillé, C.; Chanez, P.; Erjefält, J.S.; et al. Effectiveness of bronchial thermoplasty in patients with severe refractory asthma: Clinical and histopathologic correlations. J. Allergy Clin. Immunol. 2017, 139, 1176–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, I.H.; Boulet, L.-P.; Biardel, S.; Martel, S.; LaViolette, M.; Chakir, J.; Lampron, N. Long-Term Effects of Bronchial Thermoplasty on Airway Smooth Muscle and Reticular Basement Membrane Thickness in Severe Asthma. Ann. Am. Thorac. Soc. 2016, 13, 1426–1428. [Google Scholar] [CrossRef]
- Facciolongo, N.; Di Stefano, A.; Pietrini, V.; Galeone, C.; Bellanova, F.; Menzella, F.; Scichilone, N.; Piro, R.; Bajocchi, G.L.; Balbi, B.; et al. Nerve ablation after bronchial thermoplasty and sustained improvement in severe asthma. BMC Pulm. Med. 2018, 18, 29. [Google Scholar] [CrossRef]
- Cox, G.; Thomson, N.; Rubin, A.S.; Niven, R.; Corris, P.A.; Siersted, H.C.; Olivenstein, R.; Pavord, I.; McCormack, D.; Chaudhuri, R.; et al. Asthma Control during the Year after Bronchial Thermoplasty. N. Engl. J. Med. 2007, 356, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Pavord, I.D.; Cox, G.; Thomson, N.C.; Rubin, A.S.; Corris, P.A.; Niven, R.M.; Chung, K.F.; LaViolette, M.; RISA Trial Study Group. Safety and Efficacy of Bronchial Thermoplasty in Symptomatic, Severe Asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Rubin, A.S.; LaViolette, M.; Fiterman, J.; Lima, M.D.A.; Shah, P.; Fiss, E.; Olivenstein, R.; Thomson, N.; Niven, R.; et al. Effectiveness and Safety of Bronchial Thermoplasty in the Treatment of Severe Asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goorsenberg, A.W.M.; D’Hooghe, J.N.S.; Srikanthan, K.; Hacken, N.H.T.T.; Weersink, E.J.M.; Roelofs, J.J.T.H.; Kemp, S.V.; Bel, E.H.; Shah, P.L.; Annema, J.T.; et al. Bronchial Thermoplasty Induced Airway Smooth Muscle Reduction and Clinical Response in Severe Asthma. The TASMA Randomized Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; VanderStichele, C.; Jordens, P.; Deman, R.; Slabbynck, H.; Ringoet, V.; Verleden, G.; Demedts, I.K.; Verhamme, K.; Delporte, A.; et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): A multicentre randomised double-blind placebo-controlled trial. Thorax 2013, 68, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, P.G.; Yang, I.; Upham, J.; Reynolds, P.N.; Hodge, S.; James, A.L.; Jenkins, C.; Peters, M.; Marks, G.B.; Baraket, M.; et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Serisier, D.J. Risks of population antimicrobial resistance associated with chronic macrolide use for inflammatory airway diseases. Lancet Respir. Med. 2013, 1, 262–274. [Google Scholar] [CrossRef]
- McCubbrey, A.L.; Curtis, J.L. Efferocytosis and Lung Disease. Chest 2013, 143, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, C.; Gogali, A.; Bartziokas, K.; Kostikas, K. Identification and treatment of T2-low asthma in the era of biologics. ERJ Open Res. 2021, 7, 00309–02020. [Google Scholar] [CrossRef]
- Iwamoto, H.; Yokoyama, A.; Shiota, N.; Shoda, H.; Haruta, Y.; Hattori, N.; Kohno, N. Tiotropium bromide is effective for severe asthma with noneosinophilic phenotype. Eur. Respir. J. 2008, 31, 1379–1380. [Google Scholar] [CrossRef]
- Casale, T.B.; Bateman, E.D.; Vandewalker, M.; Virchow, J.C.; Schmidt, H.; Engel, M.; Moroni-Zentgraf, P.; Kerstjens, H. Tiotropium Respimat Add-on Is Efficacious in Symptomatic Asthma, Independent of T2 Phenotype. J. Allergy Clin. Immunol. Pract. 2018, 6, 923–935.e9. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Bertolini, F.; Carriero, V.; Sprio, A.E. Asthma phenotypes and endotypes. Minerva Med. 2021, 112. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Watanabe, N.; Kido, M.; Saga, K.; Akamatsu, T.; Nishio, A.; Chiba, T. Human TSLP and TLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin. Exp. Allergy 2009, 39, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Pelaia, G.; Crimi, C.; Maglio, A.; Gallelli, L.; Terracciano, R.; Vatrella, A. Tezepelumab: A Potential New Biological Therapy for Severe Refractory Asthma. Int. J. Mol. Sci. 2021, 22, 4369. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Spadaro, G.; Braile, M.; Poto, R.; Criscuolo, G.; Pahima, H.; Loffredo, S.; Levi-Schaffer, F.; Varricchi, G. Tezepelumab: A novel biological therapy for the treatment of severe uncontrolled asthma. Expert Opin. Investig. Drugs 2019, 28, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Zhang, M.; Fei, X.; Zhang, G.-Q.; Zhang, P.-Y.; Li, F.; Bao, W.-P.; Zhang, Y.-Y.; Zhou, X. Role of neutralizing anti-murine interleukin-17A monoclonal antibody on chronic ozone-induced airway inflammation in mice. Biomed. Pharmacother. 2016, 83, 247–256. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01478360?term=secukinumab&cond=Asthma&rank=1. (accessed on 15 October 2021).
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.-L. Randomized, Double-Blind, Placebo-controlled Study of Brodalumab, a Human Anti–IL-17 Receptor Monoclonal Antibody, in Moderate to Severe Asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health Clinical Center. Efficacy and Safety of BI 655066/ABBV-066 (Risankizumab) in Patients with Severe Persistent Asthma. NCT02443298. Date Last Updated: 10 April 2019. Available online: www.clinicaltrials.gov/ct2/show/NCT02443298 (accessed on 15 October 2021).
- Howarth, P.H.; Babu, K.S.; Arshad, H.S.; Lau, L.; Buckley, M.; McConnell, W.; Beckett, P.; Al Ali, M.; Chauhan, A.; Wilson, S.J.; et al. Tumour necrosis factor (TNF ) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 2005, 60, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Berry, M.A.; Hargadon, B.; Shelley, M.; Parker, D.; Shaw, D.; Green, R.H.; Bradding, P.; Brightling, C.; Wardlaw, A.; Pavord, I.D. Evidence of a Role of Tumor Necrosis Factor α in Refractory Asthma. N. Engl. J. Med. 2006, 354, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.E.; Barnes, P.J.; Bleecker, E.R.; Bousquet, J.; Busse, W.; Dahlén, S.-E.; Holgate, S.T.; Meyers, D.A.; Rabe, K.F.; Antczak, A.; et al. A Randomized, Double-blind, Placebo-controlled Study of Tumor Necrosis Factor-α Blockade in Severe Persistent Asthma. Am. J. Respir. Crit. Care Med. 2009, 179, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Pinkerton, J.W.; Essilfie, A.T.; Robertson, A.A.B.; Baines, K.J.; Brown, A.C.; Mayall, J.R.; Ali, M.K.; Starkey, M.R.; Hansbro, N.G.; et al. Role for NLRP3 Inflammasome–mediated, IL-1β–Dependent Responses in Severe, Steroid-Resistant Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Watz, H.; Uddin, M.; Pedersen, F.; Kirsten, A.; Goldmann, T.; Stellmacher, F.; Groth, E.; Larsson, B.; Böttcher, G.; Malmgren, A.; et al. Effects of the CXCR2 antagonist AZD5069 on lung neutrophil recruitment in asthma. Pulm. Pharmacol. Ther. 2017, 45, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Gaga, M.; Zervas, E.; Alagha, K.; Hargreave, F.E.; O’Byrne, P.M.; Stryszak, P.; Gann, L.; Sadeh, J.; Chanez, P.; et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: A randomized, placebo-controlled clinical trial. Clin. Exp. Allergy 2012, 42, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, P.M.; Metev, H.; Puu, M.; Richter, K.; Keen, C.; Uddin, M.; Larsson, B.; Cullberg, M.; Nair, P. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: A randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2016, 4, 797–806. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricciardolo, F.L.M.; Carriero, V.; Bertolini, F. Which Therapy for Non-Type(T)2/T2-Low Asthma. J. Pers. Med. 2022, 12, 10. https://doi.org/10.3390/jpm12010010
Ricciardolo FLM, Carriero V, Bertolini F. Which Therapy for Non-Type(T)2/T2-Low Asthma. Journal of Personalized Medicine. 2022; 12(1):10. https://doi.org/10.3390/jpm12010010
Chicago/Turabian StyleRicciardolo, Fabio L. M., Vitina Carriero, and Francesca Bertolini. 2022. "Which Therapy for Non-Type(T)2/T2-Low Asthma" Journal of Personalized Medicine 12, no. 1: 10. https://doi.org/10.3390/jpm12010010
APA StyleRicciardolo, F. L. M., Carriero, V., & Bertolini, F. (2022). Which Therapy for Non-Type(T)2/T2-Low Asthma. Journal of Personalized Medicine, 12(1), 10. https://doi.org/10.3390/jpm12010010