
Clinical, Genetic and Functional Characterization of a Novel AVPR2 Missense Mutation in a Woman with X-Linked Recessive Nephrogenic Diabetes Insipidus

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Clinical Studies
2.2. Genetic Studies
2.3. Molecular Modeling of AVPR2
2.4. Cell Culture and Transfection
2.5. cAMP and PKA Activity Assays
2.6. Western Blotting and Cell Surface Biotinylation Assay
2.7. Immunofluorescence
2.8. Statistical Analysis
3. Results
3.1. Blood and Urine Biochemical Tests
3.2. Identification of a Novel Missense Mutation in AVPR2
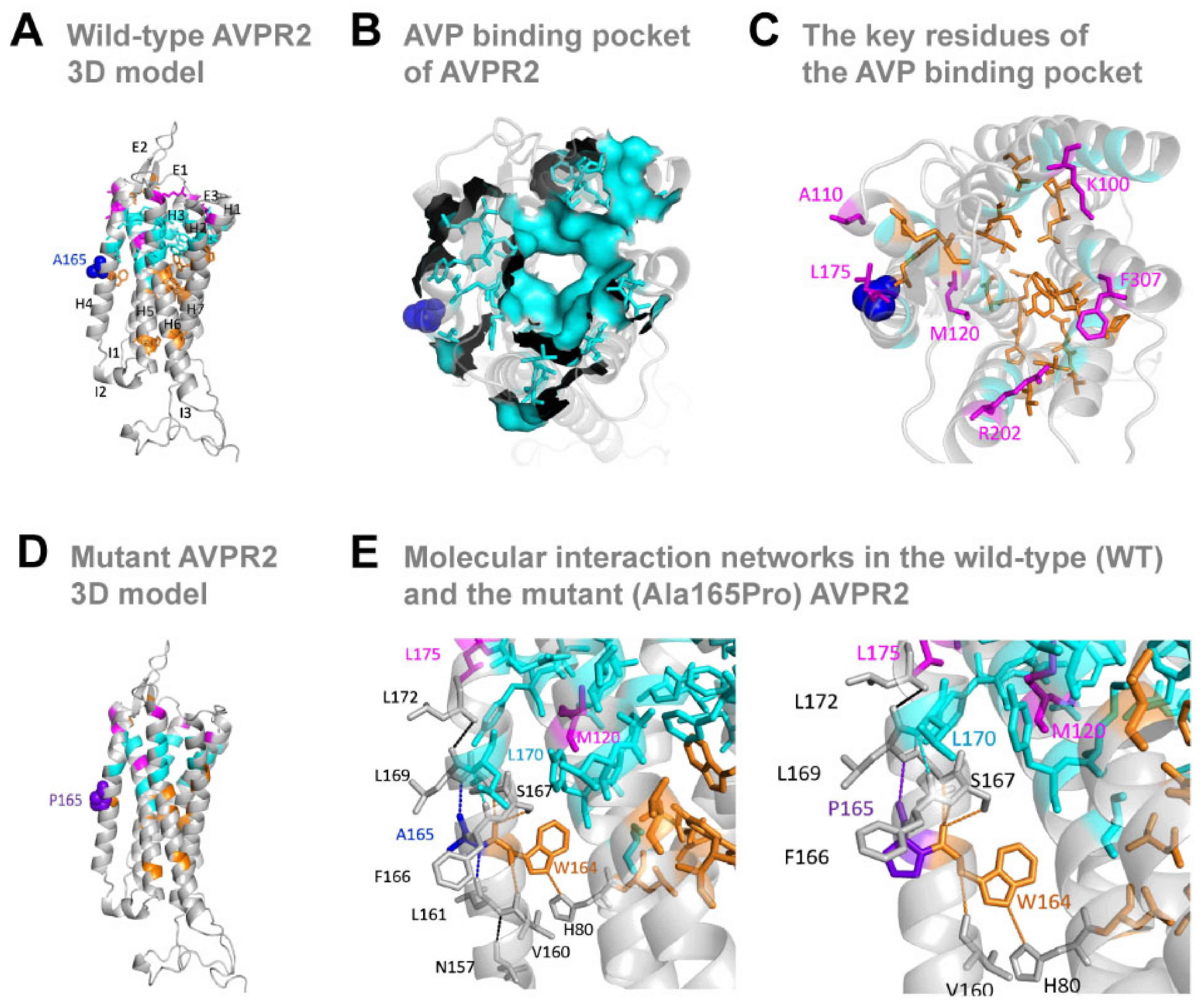
3.3. The Missense Ala165Pro Variant Disrupts AVPR2 Function
3.4. Biological Impact of the Mutant AVPR2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rondon-Berrios, H.; Berl, T. Physiology and Pathophysiology of Water Homeostasis. Front. Horm. Res. 2019, 52, 8–23. [Google Scholar] [PubMed]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G.; Verkman, A.S. Diabetes insipidus. Nat. Rev. Dis. Prim. 2019, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, C.; Uy, N.S. Nephrogenic Diabetes Insipidus. Pediatr. Clin. N. Am. 2019, 66, 227–234. [Google Scholar] [CrossRef]
- Pan, Y.; Metzenberg, A.; Das, S.; Jing, B.; Gitschier, J. Mutations in the V2 vasopressin receptor gene are associated with X-linked nephrogenic diabetes insipidus. Nat. Genet. 1992, 2, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, W.; Seibold, A.; Antaramian, A.; Lonergan, M.; Arthus, M.F.; Hendy, G.N.; Birnbaumer, M.; Bichet, D.-G. Molecular identification of the gene responsible for congenital nephrogenic diabetes insipidus. Nature 1992, 359, 233–235. [Google Scholar] [CrossRef]
- Van den Ouweland, A.M.; Dreesen, J.C.; Verdijk, M.; Knoers, N.V.; Monnens, L.A.; Rocchi, M.; van Oost, B.A. Mutations in the vasopressin type 2 receptor gene (AVPR2) associated with nephrogenic diabetes insipidus. Nat. Genet. 1992, 2, 99–102. [Google Scholar] [CrossRef]
- Bichet, D.G. Genetics in Endocrinology Pathophysiology, diagnosis and treatment of familial nephrogenic diabetes insipidus. Eur. J. Endocrinol. 2020, 183, R29–R40. [Google Scholar] [CrossRef]
- D’Alessandri-Silva, C.; Carpenter, M.; Ayoob, R.; Barcia, J.; Chishti, A.; Constantinescu, A.; Dell, K.M.; Goodwin, J.; Hashmat, S.; Iragorri, S.; et al. Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study. Front. Pediatr. 2019, 7, 550. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Ashton, E.; Iancu, D.; Arthus, M.F.; Hayes, W.; Van’t Hoff, W.; Kleta, R.; Bichet, D.G.; Bockenhauer, D. Long-term outcome in inherited nephrogenic diabetes insipidus. Clin. Kidney J. 2019, 12, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Birnbaumer, M.; Seibold, A.; Gilbert, S.; Ishido, M.; Barberis, C.; Antaramian, A.; Brabet, P.; Rosenthal, W. Molecular cloning of the receptor for human antidiuretic hormone. Nature 1992, 357, 333–335. [Google Scholar] [CrossRef]
- Seibold, A.; Brabet, P.; Rosenthal, W.; Birnbaumer, M. Structure and chromosomal localization of the human antidiuretic hormone receptor gene. Am. J. Hum. Genet. 1992, 51, 1078–1083. [Google Scholar] [PubMed]
- Spanakis, E.; Milord, E.; Gragnoli, C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: Review and missense mutation significance. J. Cell. Physiol. 2008, 217, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Beetz, R.; Rittner, G.; Bartsch, O. A female with X-linked Nephrogenic diabetes insipidus in a family with inherited central diabetes Insipidus: Case report and review of the literature. Am. J. Med. Genet. Part A 2020, 182, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichet, D.G.; Razi, M.; Lonergan, M.; Arthus, M.F.; Papukna, V.; Kortas, C.; Barjon, J.-N. Hemodynamic and coagulation responses to 1-desamino[8-D-arginine] vasopressin in patients with congenital nephrogenic diabetes insipidus. N. Engl. J. Med. 1988, 318, 881–887. [Google Scholar] [CrossRef]
- Fakhro, K.A.; Robay, A.; Rodrigues-Flores, J.L.; Mezey, J.G.; Al-Shakaki, A.A.; Chidiac, O.; Stadler, D.; Malek, J.A.; Imam, A.B.; Sheikh, A.; et al. Point of Care Exome Sequencing Reveals Allelic and Phenotypic Heterogeneity Underlying Mendelian disease in Qatar. Hum. Mol. Genet. 2019, 28, 3970–3981. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Ritchie, G.R.; Flicek, P. Computational approaches to interpreting genomic sequence variation. Genome Med. 2014, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Macion-Dazard, R.; Callahan, N.; Xu, Z.; Wu, N.; Thibonnier, M.; Shoham, M. Mapping the binding site of six nonpeptide antagonists to the human V2-renal vasopressin receptor. J. Pharmacol. Exp. Ther. 2006, 316, 564–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibonnier, M.; Graves, M.K.; Wagner, M.S.; Chatelain, N.; Soubrier, F.; Corvol, P.; Willard, H.F.; Jeunemaitre, X. Study of V(1)-vascular vasopressin receptor gene microsatellite polymorphisms in human essential hypertension. J. Mol. Cell. Cardiol. 2000, 32, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Decaux, G.; Soupart, A.; Vassart, G. Non-peptide arginine-vasopressin antagonists: The vaptans. Lancet 2008, 371, 1624–1632. [Google Scholar] [CrossRef]
- Suno, R.; Kimura, K.T.; Nakane, T.; Yamashita, K.; Wang, J.; Fujiwara, T.; Yamanaka, Y.; Im, D.; Horita, S.; Tsujimoto, H.; et al. Crystal Structures of Human Orexin 2 Receptor Bound to the Subtype-Selective Antagonist EMPA. Structure 2018, 26, 7–19.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappas, M.; Ali, A.A.E.; Bennett, K.A.; Brown, J.D.; Bucknell, S.J.; Congreve, M.; Cooke, R.M.; Cseke, G.; De Graaf, C.; Doré, A.S.; et al. Comparison of Orexin 1 and Orexin 2 Ligand Binding Modes Using X-ray Crystallography and Computational Analysis. J. Med. Chem. 2020, 63, 1528–1543. [Google Scholar] [CrossRef]
- Waltenspuhl, Y.; Schoppe, J.; Ehrenmann, J.; Kummer, L.; Pluckthun, A. Crystal structure of the human oxytocin receptor. Sci. Adv. 2020, 6, eabb5419. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef]
- Hellmann, J.; Drabek, M.; Yin, J.; Gunera, J.; Proll, T.; Kraus, F.; Langmead, C.J.; Hübner, H.; Weikert, D.; Kolb, P.; et al. Structure-based development of a subtype-selective orexin 1 receptor antagonist. Proc. Natl. Acad. Sci. USA 2020, 117, 18059–18067. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 2647. [Google Scholar] [CrossRef] [Green Version]
- Zaki, O.K.; Krishnamoorthy, N.; El Abd, H.S.; Harche, S.A.; Mattar, R.A.; Al Disi, R.S.; Nofal, M.Y.; El Bekay, R.; Ahmed, A.K.; Doss, C.G.P.; et al. Two patients with Canavan disease and structural modeling of a novel mutation. Metab. Brain Dis. 2017, 32, 171–177. [Google Scholar] [CrossRef]
- Gajendrarao, P.; Krishnamoorthy, N.; Kassem, H.; Moharem-Elgamal, S.; Cecchi, F.; Olivotto, I.; Yacoub, M.H. Molecular modeling of disease causing mutations in domain C1 of cMyBP-C. PLoS ONE 2013, 8, e59206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Tahtaoui, C.; Balestre, M.N.; Klotz, P.; Rognan, D.; Barberis, C.; Mouillac, B.; Hibert, M. Identification of the binding sites of the SR49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfydryl-reactive ligands and cysteine mutants as chemical sensors. J. Biol. Chem. 2003, 278, 40010–40019. [Google Scholar] [CrossRef] [Green Version]
- Cordes, F.S.; Bright, J.N.; Sansom, M.S. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 2002, 323, 951–960. [Google Scholar] [CrossRef]
- Grimm, C.; Cuajungco, M.P.; van Aken, A.F.; Schnee, M.; Jors, S.; Kros, C.J.; Ricci, A.J.; Heller, S. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 19583–19588. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Sangkuhl, K.; Lennert, T.; Wigger, M.; Price, D.A.; Nuuja, A.; Grüters, A.; Schultz, G.; Schöneberg, T. Aminoglycoside pretreatment partially restores the function of truncated V(2) vasopressin receptors found in patients with nephrogenic diabetes insipidus. J. Clin. Endocrinol. Metab. 2002, 87, 5247–5257. [Google Scholar] [CrossRef] [Green Version]
- Boone, M.; Deen, P.M. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflug. Arch. 2008, 456, 1005–1024. [Google Scholar] [CrossRef] [Green Version]
- Migeon, B.R. X inactivation, female mosaicism, and sex differences in renal diseases. J. Am. Soc. Nephrol. 2008, 19, 2052–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, A.M.; Sangani, G.; Miller, J.L. Proposed cause of marked vasopressin resistance in a female with an X-linked recessive V2 receptor abnormality. J. Clin. Endocrinol. Metab. 1995, 80, 1184–1186. [Google Scholar] [PubMed]
- Oksche, A.; Schulein, R.; Rutz, C.; Liebenhoff, U.; Dickson, J.; Muller, H.; Birnbaumer, M.; Rosenthal, W. Vasopressin V2 receptor mutants that cause X-linked nephrogenic diabetes insipidus: Analysis of expression, processing, and function. Mol. Pharmacol. 1996, 50, 820–828. [Google Scholar]
- Nejsum, L.N.; Christensen, T.M.; Robben, J.H.; Milligan, G.; Deen, P.M.; Bichet, D.G.; Levin, K. Novel mutation in the AVPR2 gene in a Danish male with nephrogenic diabetes insipidus caused by ER retention and subsequent lysosomal degradation of the mutant receptor. NDT Plus 2011, 4, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Boselt, I.; Tramma, D.; Kalamitsou, S.; Niemeyer, T.; Nykanen, P.; Graf, K.J.; Krude, H.; Marenzi, K.S.; Di Candia, S.; Schöneberg, T.; et al. Functional characterization of novel loss-of-function mutations in the vasopressin type 2 receptor gene causing nephrogenic diabetes insipidus. Nephrol. Dial. Transplant. 2012, 27, 1521–1528. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, E.; Greenbaum, L.A.; Rochdi, D.; Abrol, R.; Goddard, W.A.; Bichet, D.G., III; Bouvier, M. Identification and characterization of an activating F229V substitution in the V2 vasopressin receptor in an infant with NSIAD. J. Am. Soc. Nephrol. 2012, 23, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Tiulpakov, A.; White, C.W.; Abhayawardana, R.S.; See, H.B.; Chan, A.S.; Seeber, R.M.; Heng, J.I.; Dedov, I.; Pavlos, N.J.; Pfleger, K.D. Mutations of Vasopressin Receptor 2 Including Novel L312S Have Differential Effects on Trafficking. Mol. Endocrinol. 2016, 30, 889–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, J.P.; Salahpour, A.; Laperriere, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petäjä-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouley, R.; Pastor-Soler, N.; Cohen, O.; McLaughlin, M.; Breton, S.; Brown, D. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra). Am. J. Physiol. Renal Physiol. 2005, 288, F1103–F11012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.L.; Sandoval, P.C.; Pisitkun, T.; Knepper, M.A.; Hoffert, J.D. Vasopressin inhibits apoptosis in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 2013, 304, F177–F188. [Google Scholar] [CrossRef] [Green Version]
- Goel, M.; Zuo, C.D.; Schilling, W.P. Role of cAMP/PKA signaling cascade in vasopressin-induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am. J. Physiol. Renal Physiol. 2010, 298, F988–F996. [Google Scholar] [CrossRef] [Green Version]
- Prosperi, F.; Suzumoto, Y.; Marzuillo, P.; Costanzo, V.; Jelen, S.; Iervolino, A.; Guarino, S.; La Manna, A.; Del Giudice, E.M.; Perna, A.F.; et al. Characterization of five novel vasopressin V2 receptor mutants causing nephrogenic diabetes insipidus reveals a role of tolvaptan for M272R-V2R mutation. Sci. Rep. 2020, 10, 16383. [Google Scholar] [CrossRef]
- Zhang, J.; Bui, T.N.; Xiang, J.; Lin, A. Cyclic AMP inhibits p38 activation via CREB-induced dynein light chain. Mol. Cell. Biol. 2006, 26, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Nedvetsky, P.I.; Tabor, V.; Tamma, G.; Beulshausen, S.; Skroblin, P.; Kirschner, A.; Mutig, K.; Boltzen, M.; Petrucci, O.; Vossenkämper, A.; et al. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J. Am. Soc. Nephrol. 2010, 21, 1645–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, B.M.; Marples, D.; Jensen, U.B.; Frokiaer, J.; Sheikh-Hamad, D.; Knepper, M.; Nielsen, S. Acute effects of vasopressin V2-receptor antagonist on kidney AQP2 expression and subcellular distribution. Am. J. Physiol. 1998, 275, F285–F297. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selvaraj, S.; Rodrigues, D.; Krishnamoorthy, N.; Fakhro, K.A.; Saraiva, L.R.; Lemos, M.C. Clinical, Genetic and Functional Characterization of a Novel AVPR2 Missense Mutation in a Woman with X-Linked Recessive Nephrogenic Diabetes Insipidus. J. Pers. Med. 2022, 12, 118. https://doi.org/10.3390/jpm12010118
Selvaraj S, Rodrigues D, Krishnamoorthy N, Fakhro KA, Saraiva LR, Lemos MC. Clinical, Genetic and Functional Characterization of a Novel AVPR2 Missense Mutation in a Woman with X-Linked Recessive Nephrogenic Diabetes Insipidus. Journal of Personalized Medicine. 2022; 12(1):118. https://doi.org/10.3390/jpm12010118
Chicago/Turabian StyleSelvaraj, Senthil, Dírcea Rodrigues, Navaneethakrishnan Krishnamoorthy, Khalid A. Fakhro, Luís R. Saraiva, and Manuel C. Lemos. 2022. "Clinical, Genetic and Functional Characterization of a Novel AVPR2 Missense Mutation in a Woman with X-Linked Recessive Nephrogenic Diabetes Insipidus" Journal of Personalized Medicine 12, no. 1: 118. https://doi.org/10.3390/jpm12010118
APA StyleSelvaraj, S., Rodrigues, D., Krishnamoorthy, N., Fakhro, K. A., Saraiva, L. R., & Lemos, M. C. (2022). Clinical, Genetic and Functional Characterization of a Novel AVPR2 Missense Mutation in a Woman with X-Linked Recessive Nephrogenic Diabetes Insipidus. Journal of Personalized Medicine, 12(1), 118. https://doi.org/10.3390/jpm12010118