
Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
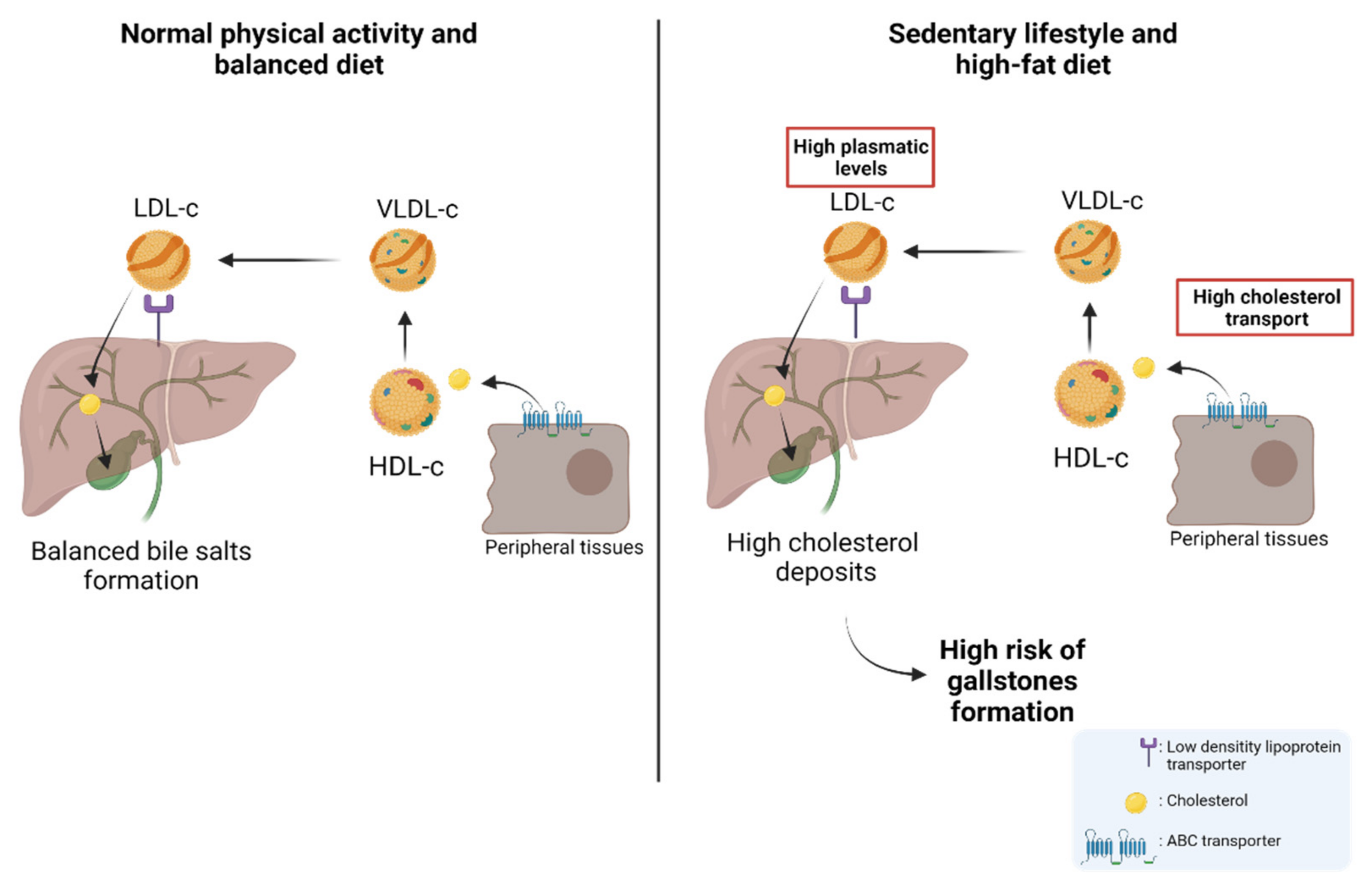
:1. Introduction
2. Search and Selection of Literature
3. Inflammation in Gallbladder Carcinogenesis
4. Nutritional and Lifestyle Aspects That Increase Susceptibility to Gallbladder Carcinogenesis
5. Infections and Chronic Inflammatory Diseases
5.1. Infections Related to Gallbladder Carcinogenesis
5.2. Immune Inflammatory Diseases Involved in Gallbladder Carcinogenesis
6. Consumption of Alcohol and Tobacco as a Risk Factor for Gallbladder Carcinogenesis
7. Exposure to Aflatoxins and Other Elements and Compounds as Risk Factors for Gallbladder Carcinogenesis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin Epidemiol 2014, 6, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Alvi, A.R.; Siddiqui, N.A.; Zafar, H. Risk factors of gallbladder cancer in Karachi-a case-control study. World J. Surg. Oncol. 2011, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa, I.; Ibacache, G.; Roa, J.; Araya, J.; de Aretxabala, X.; Muñoz, S. Gallstones and gallbladder cancer-volume and weight of gallstones are associated with gallbladder cancer: A case-control study. J. Surg. Oncol. 2006, 93, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Gao, Y.-T.; Han, T.-Q.; Rashid, A.; Sakoda, L.C.; Wang, B.-S.; Shen, M.-C.; Zhang, B.-H.; Niwa, S.; Chen, J.; et al. Gallstones and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2007, 97, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Bizama, C.; García, P.; Ferreccio, C.; Javle, M.; Miquel, J.F.; Koshiol, J.; Roa, J.C. The inflammatory inception of gallbladder cancer. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2016, 1865, 245–254. [Google Scholar] [CrossRef]
- Stokes, C.S.; Krawczyk, M.; Lammert, F. Gallstones: Environment, Lifestyle and Genes. Dig. Dis. 2011, 29, 191–201. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronicSalmonella typhicarrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750. [Google Scholar] [CrossRef]
- Zhu, Q.; Sun, X.; Ji, X.; Zhu, L.; Xu, J.; Wang, C.; Zhang, C.; Xue, F.; Liu, Y. The association between gallstones and metabolic syndrome in urban Han Chinese: A longitudinal cohort study. Sci. Rep. 2016, 6, 29937. [Google Scholar] [CrossRef] [Green Version]
- van Erp, L.W.; Cunningham, M.; Narasimman, M.; Ali, H.A.; Jhaveri, K.; Drenth, J.P.H.; Janssen, H.L.A.; Levy, C.; Hirschfield, G.M.; Hansen, B.E.; et al. Risk of gallbladder cancer in patients with primary sclerosing cholangitis and radiographically detected gallbladder polyps. Liver Int. 2020, 40, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Xuan, Y.; Li, X.; Crawford, W.J.; Yuan, Z.; Chen, Z.; Brooks, A.; Song, Y.; Wang, H.; Liang, X.; et al. Effect of metabolic syndrome components on the risk of malignancy in patients with gallbladder lesions. J. Cancer 2021, 12, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Yan, S.; Wang, B.; Shen, F.; Cao, H.; Fan, J.; Wang, Y. Type 2 diabetes mellitus and risk of gallbladder cancer: A systematic review and meta-analysis of observational studies. Diabetes/Metab. Res. Rev. 2015, 32, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wenbin, D.; Zhuo, C.; Zhibing, M.; Chen, Z.; Ruifan, Y.; Jie, J.; Cheng, Q.; Zhenming, G. The effect of smoking on the risk of gallbladder cancer: A meta-analysis of observational studies. Eur. J. Gastroenterol. Hepatol. 2013, 25, 373–379. [Google Scholar] [CrossRef]
- Yagyu, K.; Kikuchi, S.; Obata, Y.; Lin, Y.; Ishibashi, T.; Kurosawa, M.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Cigarette smoking, alcohol drinking and the risk of gallbladder cancer death: A prospective cohort study in Japan. Int. J. Cancer 2008, 122, 924–929. [Google Scholar] [CrossRef] [PubMed]
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.-O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; De Gonzalez, A.B.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Al Manasra, A.R.A.; Jadallah, K.; Aleshawi, A.; Al-Omari, M.; Elheis, M.; Reyad, A.; Fataftah, J.; Al-Domaidat, H. Intractable Biliary Candidiasis in Patients with Obstructive Jaundice and Regional Malignancy: A Retrospective Case Series. Clin. Exp. Gastroenterol. 2021, 14, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Giovannucci, E.L.; Wolk, A. Sweetened Beverage Consumption and Risk of Biliary Tract and Gallbladder Cancer in a Prospective Study. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Borena, W.; Edlinger, M.; Bjørge, T.; Häggström, C.; Lindkvist, B.; Nagel, G.; Engeland, A.; Stocks, T.; Strohmaier, S.; Manjer, J.; et al. A prospective study on metabolic risk factors and gallbladder cancer in the metabolic syndrome and cancer (Me-Can) collaborative study. PLoS ONE 2014, 9, e89368. [Google Scholar] [CrossRef] [Green Version]
- Hayat, S.; Hassan, Z.; Changazi, S.H.; Zahra, A.; Noman, M.; Zain Ul Abdin, M.; Javed, H.; Ans, A.H. Comparative analysis of serum lipid profiles in patients with and without gallstones: A prospective cross-sectional study. Ann. Med. Surg. 2019, 42, 11–13. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Tan, W.; Gao, M.; Liu, N.; Zhang, G.; Xu, T.; Cui, W. Body Mass Index and Risk of Gallbladder Cancer: Systematic Review and Meta-Analysis of Observational Studies. Nutrients 2015, 7, 8321–8334. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Gao, Y.; Wang, Y. Helicobacter species infection may be associated with cholangiocarcinoma: A meta-analysis. Int J Clin. Pract. 2014, 68, 262–270. [Google Scholar] [CrossRef]
- Chen, Z.X.; Peng, X.T.; Tan, L.; Zhai, G.Q.; Chen, G.; Gan, T.Q.; Li, J.J. EBV as a potential risk factor for hepatobiliary system cancer: A meta-analysis with 918 cases. Pathol. Res. Pract. 2019, 215, 278–285. [Google Scholar] [CrossRef]
- O’Keeffe, L.M.; Taylor, G.; Huxley, R.R.; Mitchell, P.; Woodward, M.; Peters, S.A.E. Smoking as a risk factor for lung cancer in women and men: A systematic review and meta-analysis. BMJ Open 2018, 8, e021611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, A.J.; Kashef Hamadani, B.H.; Kumaran, E.A.P.; Rao, P.; Longbottom, J.; Harriss, E.; Moore, C.E.; Dunachie, S.; Basnyat, B.; Baker, S.; et al. Drug-resistant enteric fever worldwide, 1990 to 2018: A systematic review and meta-analysis. BMC Med. 2020, 18, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.; Shukla, V.K. Diet and gallbladder cancer: A case-control study. Eur. J. Cancer Prev. 2002, 11, 365–368. [Google Scholar] [CrossRef]
- Mhatre, S.; Wang, Z.; Nagrani, R.; Badwe, R.; Chiplunkar, S.; Mittal, B.; Yadav, S.; Zhang, H.; Chung, C.C.; Patil, P.; et al. Common genetic variation and risk of gallbladder cancer in India: A case-control genome-wide association study. Lancet Oncol. 2017, 18, 535–544. [Google Scholar] [CrossRef]
- Strom, B.L.; Soloway, R.D.; Rios-Dalenz, J.L.; Rodriguez-Martinez, H.A.; West, S.L.; Kinman, J.L.; Polansky, M.; Berlin, J.A. Risk factors for gallbladder cancer. An international collaborative case-control study. Cancer 1995, 76, 1747–1756. [Google Scholar] [CrossRef]
- Lee, B.S.; Park, E.C.; Park, S.W.; Nam, C.M.; Roh, J. Hepatitis B virus infection, diabetes mellitus, and their synergism for cholangiocarcinoma development: A case-control study in Korea. World J. Gastroenterol. 2015, 21, 502–510. [Google Scholar] [CrossRef]
- Torabi Sagvand, B.; Edwards, K.; Shen, B. Frequency, Risk Factors, and Outcome of Gallbladder Polyps in Patients With Primary Sclerosing Cholangitis: A Case-Control Study. Hepatol. Commun. 2018, 2, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.C.; Chien, W.C.; Hu, J.M.; Tzeng, N.S.; Chung, C.H.; Pu, T.W.; Hsiao, C.W.; Chen, C.Y. Risk of colorectal cancer in patients with alcoholism: A nationwide, population-based nested case-control study. PLoS ONE 2020, 15, e0232740. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.K.; Prakash, A.; Tripathi, B.D.; Reddy, D.C.; Singh, S. Biliary heavy metal concentrations in carcinoma of the gall bladder: Case-control study. BMJ 1998, 317, 1288–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farioli, A.; Straif, K.; Brandi, G.; Curti, S.; Kjaerheim, K.; Martinsen, J.I.; Sparen, P.; Tryggvadottir, L.; Weiderpass, E.; Biasco, G.; et al. Occupational exposure to asbestos and risk of cholangiocarcinoma: A population-based case-control study in four Nordic countries. Occup. Environ. Med. 2018, 75, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Barahona Ponce, C.; Scherer, D.; Boekstegers, F.; Garate-Calderon, V.; Jenab, M.; Aleksandrova, K.; Katzke, V.; Weiderpass, E.; Bonet, C.; Moradi, T.; et al. Arsenic and gallbladder cancer risk: Mendelian randomization analysis of European prospective data. Int. J. Cancer 2020, 146, 2648–2650. [Google Scholar] [CrossRef] [PubMed]
- Barahona Ponce, C.; Scherer, D.; Brinster, R.; Boekstegers, F.; Marcelain, K.; Gárate-Calderón, V.; Müller, B.; de Toro, G.; Retamales, J.; Barajas, O.; et al. Gallstones, Body Mass Index, C-Reactive Protein, and Gallbladder Cancer: Mendelian Randomization Analysis of Chilean and European Genotype Data. Hepatology 2021, 73, 1783–1796. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Weiss, U. Inflammation. Nature 2008, 454, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys 2003, 417, 3–11. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Loyher, P.L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.S.; Zhou, L.S.; Han, Y.; Zhu, A.J.; Sun, X.J.; Yang, Y.J. Expression of tumor necrosis factor and its receptor in gallstone and gallbladder carcinoma tissue. Hepatobiliary Pancreat Dis. Int. 2004, 3, 448–452. [Google Scholar]
- Nilsson, B.; Delbro, D.; Hedin, L.; Friman, S.; Andius, S.; Svanvik, J. Role of cyclooxygenase-2 for fluid secretion by the inflamed gallbladder mucosa. J. Gastrointest. Surg. 1998, 2, 269–277. [Google Scholar] [CrossRef]
- Longo, W.E.; Panesar, N.; Mazuski, J.E.; Kaminski, D. Synthetic pathways of gallbladder mucosal prostanoids: The role of cyclooxygenase-1 and 2. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 77–85. [Google Scholar] [CrossRef]
- Thornell, E.; Jansson, R.; Kral, J.G.; Svanvik, J. Inhibition of prostaglandin synthesis as a treatment for biliary pain. Lancet 1979, 1, 584. [Google Scholar] [CrossRef]
- Babb, R.R. Managing gallbladder disease with prostaglandin inhibitors. Postgrad. Med. 1993, 94, 127–130. [Google Scholar] [CrossRef]
- Fumino, S.; Tokiwa, K.; Ono, S.; Iwai, N. Cyclooxygenase-2 expression in the gallbladder of patients with anomalous arrangement of the pancreaticobiliary duct. J. Pediatr. Surg. 2003, 38, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Fumino, S.; Tokiwa, K.; Katoh, T.; Ono, S.; Iwai, N. New insight into bile flow dynamics in anomalous arrangement of the pancreaticobiliary duct. Br. J. Surg. 2002, 89, 865–869. [Google Scholar] [CrossRef]
- Tokiwa, K.; Iwai, N. Early mucosal changes of the gallbladder in patients with anomalous arrangement of the pancreaticobiliary duct. Gastroenterology 1996, 110, 1614–1618. [Google Scholar] [CrossRef]
- Sasatomi, E.; Tokunaga, O.; Miyazaki, K. Precancerous conditions of gallbladder carcinoma: Overview of histopathologic characteristics and molecular genetic findings. J. Hepatobiliary Pancreat. Surg. 2000, 7, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Seretis, C.; Lagoudianakis, E.; Gemenetzis, G.; Seretis, F.; Pappas, A.; Gourgiotis, S. Metaplastic changes in chronic cholecystitis: Implications for early diagnosis and surgical intervention to prevent the gallbladder metaplasia-dysplasia-carcinoma sequence. J. Clin. Med. Res. 2014, 6, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Tsuchida, A.; Iwao, T.; Eguchi, N.; Sasaki, T.; Morinaka, K.; Matsubara, K.; Kawasaki, Y.; Yamamoto, S.; Kajiyama, G. Gene mutations of K-ras in gallbladder mucosae and gallbladder carcinoma with an anomalous junction of the pancreaticobiliary duct. Am. J. Gastroenterol. 1999, 94, 1638–1642. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Rashid, A.; Churi, C.; Kar, S.; Zuo, M.; Eterovic, A.K.; Nogueras-Gonzalez, G.M.; Janku, F.; Shroff, R.T.; Aloia, T.A.; et al. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum. Pathol. 2014, 45, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Tsuji, T.; Miyama, A.; Yamaguchi, H.; Funabiki, T. Mutagenicity of bile and pancreatic juice from patients with pancreatico-biliary maljunction. Hepatogastroenterology 1995, 42, 113–116. [Google Scholar]
- Yamagiwa, H.; Tomiyama, H. Intestinal metaplasia-dysplasia-carcinoma sequence of the gallbladder. Pathol. Int. 1986, 36, 989–997. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; Kumari, N.; Krishnani, N.; Rastogi, N. Mutational frequency of. Ecancermedicalscience 2017, 11, 757. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.K.; Chetri, K.; Pandey, U.B.; Kapoor, V.K.; Mittal, B.; Choudhuri, G. Mutational spectrum of K-ras oncogene among Indian patients with gallbladder cancer. J. Gastroenterol. Hepatol. 2004, 19, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltrán, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar]
- Sharma, A.; Sharma, K.L.; Gupta, A.; Yadav, A.; Kumar, A. Gallbladder cancer epidemiology, pathogenesis and molecular genetics: Recent update. World J. Gastroenterol. 2017, 23, 3978–3998. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.R.; Creasy, J.M.; Goldman, D.A.; Gönen, M.; Kandoth, C.; Kundra, R.; Solit, D.B.; Askan, G.; Klimstra, D.S.; Basturk, O.; et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer 2019, 125, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.; Pimentel, F.; Gazdar, A.F.; Wistuba, I.I.; Miquel, J.F. TP53 abnormalities are frequent and early events in the sequential pathogenesis of gallbladder carcinoma. Ann. Hepatol. 2005, 4, 192–199. [Google Scholar] [CrossRef]
- Koda, M.; Yashima, K.; Kawaguchi, K.; Andachi, H.; Hosoda, A.; Shiota, G.; Ito, H.; Murawaki, Y. Expression of Fhit, Mlh1, and P53 protein in human gallbladder carcinoma. Cancer Lett. 2003, 199, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Brägelmann, J.; Barahona Ponce, C.; Marcelain, K.; Roessler, S.; Goeppert, B.; Gallegos, I.; Colombo, A.; Sanhueza, V.; Morales, E.; Rivera, M.T.; et al. Epigenome-Wide Analysis of Methylation Changes in the Sequence of Gallstone Disease, Dysplasia, and Gallbladder Cancer. Hepatology 2021, 73, 2293–2310. [Google Scholar] [CrossRef]
- Jain, K.; Mohapatra, T.; Das, P.; Misra, M.C.; Gupta, S.D.; Ghosh, M.; Kabra, M.; Bansal, V.K.; Kumar, S.; Sreenivas, V.; et al. Sequential occurrence of preneoplastic lesions and accumulation of loss of heterozygosity in patients with gallbladder stones suggest causal association with gallbladder cancer. Ann. Surg. 2014, 260, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, I.I.; Ashfaq, R.; Maitra, A.; Alvarez, H.; Riquelme, E.; Gazdar, A.F. Fragile histidine triad gene abnormalities in the pathogenesis of gallbladder carcinoma. Am. J. Pathol. 2002, 160, 2073–2079. [Google Scholar] [CrossRef] [Green Version]
- Roa, I.; Garcia, H.; Game, A.; de Toro, G.; de Aretxabala, X.; Javle, M. Somatic Mutations of PI3K in Early and Advanced Gallbladder Cancer: Additional Options for an Orphan Cancer. J. Mol. Diagn. 2016, 18, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, Z. Expression of phospho-ERK1/2 and PI3-K in benign and malignant gallbladder lesions and its clinical and pathological correlations. J. Exp. Clin. Cancer Res. 2009, 28, 65. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, J.A.; Riquelme, I.; Sagredo, E.A.; Rosa, L.; García, P.; Bizama, C.; Apud-Bell, M.; Leal, P.; Weber, H.; Benavente, F.; et al. Mucin 5B, carbonic anhydrase 9 and claudin 18 are potential theranostic markers of gallbladder carcinoma. Histopathology 2019, 74, 597–607. [Google Scholar] [CrossRef]
- Mishra, S.K.; Kumari, N.; Krishnani, N. Molecular pathogenesis of gallbladder cancer: An update. Mutat. Res. 2019, 816–818, 111674. [Google Scholar] [CrossRef]
- Salazar, M.; Ituarte, C.; Abriata, M.G.; Santoro, F.; Arroyo, G. Gallbladder cancer in South America: Epidemiology and prevention. Chin. Clin. Oncol. 2019, 8, 32. [Google Scholar] [CrossRef]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.A.; Schröder, M.S.; Sreenivasamurthy, S.K.; Kim, S.W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, A.; Yamauchi, Y.; Hirohashi, S. p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int. J. Cancer 1996, 69, 28–33. [Google Scholar] [CrossRef]
- Shiao, Y.H.; Rugge, M.; Correa, P.; Lehmann, H.P.; Scheer, W.D. p53 alteration in gastric precancerous lesions. Am. J. Pathol. 1994, 144, 511–517. [Google Scholar] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Sozzi, G.; Miozzo, M.; Donghi, R.; Pilotti, S.; Cariani, C.T.; Pastorino, U.; Della Porta, G.; Pierotti, M.A. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res. 1992, 52, 6079–6082. [Google Scholar]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A Tale of Two Stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Portincasa, P. Recent advances in understanding and managing cholesterol gallstones. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Skoumas, J.; Pitsavos, C.; Panagiotakos, D.B.; Chrysohoou, C.; Zeimbekis, A.; Papaioannou, I.; Toutouza, M.; Toutouzas, P.; Stefanadis, C. Physical activity, high density lipoprotein cholesterol and other lipids levels, in men and women from the ATTICA study. Lipids Health Dis. 2003, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Chen, J.; Shen, M.C.; Han, T.Q.; Wang, B.S.; Gao, Y.T. Body size and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2008, 99, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everson, G.T.; McKinley, C.; Kern, F. Mechanisms of gallstone formation in women. Effects of exogenous estrogen (Premarin) and dietary cholesterol on hepatic lipid metabolism. J. Clin. Invest. 1991, 87, 237–246. [Google Scholar] [CrossRef]
- Pinto Pereira, S.M.; Ki, M.; Power, C. Sedentary behaviour and biomarkers for cardiovascular disease and diabetes in mid-life: The role of television-viewing and sitting at work. PLoS ONE 2012, 7, e31132. [Google Scholar] [CrossRef]
- Crichton, G.E.; Alkerwi, A. Physical activity, sedentary behavior time and lipid levels in the Observation of Cardiovascular Risk Factors in Luxembourg study. Lipids Health Dis. 2015, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [Green Version]
- Van Erpecum, K.J. Pathogenesis of cholesterol and pigment gallstones: An update. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 281–287. [Google Scholar] [CrossRef]
- Rebholz, C.; Krawczyk, M.; Lammert, F. Genetics of gallstone disease. Eur. J. Clin. Invest. 2018, 48, e12935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Chang, Y.; Yun, K.E.; Jung, H.S.; Shin, J.H.; Shin, H. Gallstones and the Risk of Gallbladder Cancer Mortality: A Cohort Study. Am. J. Gastroenterol. 2016, 111, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Stinton, L.M.; Shaffer, E.A. Epidemiology of gallbladder disease: Cholelithiasis and cancer. Gut Liver 2012, 6, 172–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, X.; Vaisman, B.; Amar, M.; Sethi, A.A.; Remaley, A.T. Lecithin: Cholesterol acyltransferase--from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Masson, D.; Jiang, X.C.; Lagrost, L.; Tall, A.R. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 2009, 50, S201–S206. [Google Scholar] [CrossRef] [Green Version]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Rader, D.J. Regulation of reverse cholesterol transport and clinical implications. Am. J. Cardiol. 2003, 92, 42J–49J. [Google Scholar] [CrossRef]
- Atamanalp, S.S.; Keles, M.S.; Atamanalp, R.S.; Acemoglu, H.; Laloglu, E. The effects of serum cholesterol, LDL, and HDL levels on gallstone cholesterol concentration. Pak. J. Med. Sci. 2013, 29, 187–190. [Google Scholar] [CrossRef]
- Wang, J.; Shen, S.; Wang, B.; Ni, X.; Liu, H.; Yu, R.; Suo, T. Serum lipid levels are the risk factors of gallbladder stones: A population-based study in China. Lipids Health Dis. 2020, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Reshetnyak, V.I. Concept of the pathogenesis and treatment of cholelithiasis. World J. Hepatol. 2012, 4, 18–34. [Google Scholar] [CrossRef]
- Shrikhande, S.V.; Barreto, S.G.; Singh, S.; Udwadia, T.E.; Agarwal, A.K. Cholelithiasis in gallbladder cancer: Coincidence, cofactor, or cause! Eur. J. Surg. Oncol. 2010, 36, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.I.; Fang, S.; Kim, K.P.; Ko, Y.; Kim, H.; Oh, J.; Hong, G.Y.; Lee, S.Y.; Kim, J.M.; Noh, I.; et al. Combination treatment with n-3 polyunsaturated fatty acids and ursodeoxycholic acid dissolves cholesterol gallstones in mice. Sci. Rep. 2019, 9, 12740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, K.J.; Rao, V.P.; Ge, Z.; Rogers, A.B.; Oura, T.J.; Carey, M.C.; Fox, J.G. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology 2007, 133, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Rosa, L.; Lobos-González, L.; Muñoz-Durango, N.; García, P.; Bizama, C.; Gómez, N.; González, X.; Wichmann, I.A.; Saavedra, N.; Guevara, F.; et al. Evaluation of the chemopreventive potentials of ezetimibe and aspirin in a novel mouse model of gallbladder preneoplasia. Mol. Oncol. 2020, 14, 2834–2852. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, B.; Nausch, B.; Zane, E.A.; Leonard, M.R.; Balemba, O.B.; Bartoo, A.C.; Wilcox, R.; Nelson, M.T.; Carey, M.C.; Mawe, G.M. Disruption of gallbladder smooth muscle function is an early feature in the development of cholesterol gallstone disease. Neurogastroenterol. Motil. 2012, 24, e313–e324. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Fushimi, K.; Yabuki, Y.; Maru, Y.; Hasegawa, S.; Matsuura, T.; Kurotaki, D.; Suzuki, A.; Kobayashi, N.; Yoneda, M.; et al. Precision modeling of gall bladder cancer patients in mice based on orthotopic implantation of organoid-derived tumor buds. Oncogenesis 2021, 10, 33. [Google Scholar] [CrossRef]
- Liu, Z.; Kemp, T.J.; Gao, Y.T.; Corbel, A.; McGee, E.E.; Wang, B.; Shen, M.C.; Rashid, A.; Hsing, A.W.; Hildesheim, A.; et al. Association of circulating inflammation proteins and gallstone disease. J. Gastroenterol. Hepatol. 2018, 33, 1920–1924. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Kim, O.H.; Lee, S.C.; Kim, K.H.; Hong, H.E.; Seo, H.; Choi, H.J.; Kim, S.J. Serum level of visfatin can reflect the severity of inflammation in patients with acute cholecystitis. Ann. Surg. Treat. Res. 2020, 99, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Grünhage, F.; Acalovschi, M.; Tirziu, S.; Walier, M.; Wienker, T.F.; Ciocan, A.; Mosteanu, O.; Sauerbruch, T.; Lammert, F. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology 2007, 46, 793–801. [Google Scholar] [CrossRef]
- Small, D.M. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci USA 2003, 100, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Kampen, O.; Buch, S.; Nothnagel, M.; Azocar, L.; Molina, H.; Brosch, M.; Erhart, W.; von Schönfels, W.; Egberts, J.; Seeger, M.; et al. Genetic and functional identification of the likely causative variant for cholesterol gallstone disease at the ABCG5/8 lithogenic locus. Hepatology 2013, 57, 2407–2417. [Google Scholar] [CrossRef]
- Bustos, B.I.; Pérez-Palma, E.; Buch, S.; Azócar, L.; Riveras, E.; Ugarte, G.D.; Toliat, M.; Nürnberg, P.; Lieb, W.; Franke, A.; et al. Variants in ABCG8 and TRAF3 genes confer risk for gallstone disease in admixed Latinos with Mapuche Native American ancestry. Sci. Rep. 2019, 9, 772. [Google Scholar] [CrossRef]
- Jackson, S.S.; Van De Wyngard, V.; Pfeiffer, R.M.; Cook, P.; Hildesheim, A.; Pinto, L.A.; Jackson, S.H.; Choi, K.; Verdugo, R.A.; Cuevas, M.; et al. Inflammatory profiles in Chilean Mapuche and non-Mapuche women with gallstones at risk of developing gallbladder cancer. Sci. Rep. 2021, 11, 3686. [Google Scholar] [CrossRef]
- Delaunay, J.L.; Durand-Schneider, A.M.; Dossier, C.; Falguières, T.; Gautherot, J.; Davit-Spraul, A.; Aït-Slimane, T.; Housset, C.; Jacquemin, E.; Maurice, M. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016, 63, 1620–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakeeb, A.; Comuzzie, A.G.; Martin, L.; Sonnenberg, G.E.; Swartz-Basile, D.; Kissebah, A.H.; Pitt, H.A. Gallstones: Genetics versus environment. Ann. Surg. 2002, 235, 842–849. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Hermelin, B.; Boelle, P.Y.; Parc, R.; Taboury, J.; Poupon, R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003, 125, 452–459. [Google Scholar] [CrossRef]
- Jansen, P.L.; Strautnieks, S.S.; Jacquemin, E.; Hadchouel, M.; Sokal, E.M.; Hooiveld, G.J.; Koning, J.H.; De Jager-Krikken, A.; Kuipers, F.; Stellaard, F.; et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology 1999, 117, 1370–1379. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Miyake, J.H.; Duong-Polk, X.T.; Taylor, J.M.; Du, E.Z.; Castellani, L.W.; Lusis, A.J.; Davis, R.A. Transgenic expression of cholesterol-7-alpha-hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Lancellotti, S.; Zaffanello, M.; Di Leo, E.; Costa, L.; Lonardo, A.; Tarugi, P. Pediatric gallstone disease in familial hypobetalipoproteinemia. J. Hepatol. 2005, 43, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Holicky, E.L.; Ulrich, C.D.; Wieben, E.D. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 1995, 109, 1375–1380. [Google Scholar] [CrossRef]
- Wang, D.Q.; Schmitz, F.; Kopin, A.S.; Carey, M.C. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J. Clin. Invest. 2004, 114, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, K.; Srivastava, A.; Kumar, A.; Mittal, B. Significant association between toll-like receptor gene polymorphisms and gallbladder cancer. Liver Int. 2010, 30, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Pandey, S.N.; Choudhuri, G.; Mittal, B. IL-1 gene polymorphisms and genetic susceptibility of gallbladder cancer in a north Indian population. Cancer Genet. Cytogenet. 2008, 186, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Andreotti, G.; Chen, J.; Wang, B.S.; Shen, M.C.; Chen, B.E.; Rosenberg, P.S.; Zhang, M.; et al. Variants in inflammation genes and the risk of biliary tract cancers and stones: A population-based study in China. Cancer Res. 2008, 68, 6442–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Pandey, S.N.; Choudhuri, G.; Mittal, B. CCR5 Delta32 polymorphism: Associated with gallbladder cancer susceptibility. Scand. J. Immunol. 2008, 67, 516–522. [Google Scholar] [CrossRef]
- Cha, P.C.; Zembutsu, H.; Takahashi, A.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies SNP in DCC is associated with gallbladder cancer in the Japanese population. J. Hum. Genet. 2012, 57, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.E.; Berger, D.L. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery 2001, 129, 699–703. [Google Scholar] [CrossRef]
- Morimoto, M.; Matsuo, T.; Mori, N. Management of Porcelain Gallbladder, Its Risk Factors, and Complications: A Review. Diagnostics 2021, 11, 1073. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.; Zakko, A.; Petrov, J.C.; Kumar, P.; Duffy, A.J.; Muniraj, T. Gallbladder Disorders: A Comprehensive Review. Dis. Mon. 2021, 67, 101130. [Google Scholar] [CrossRef]
- Berger, M.; Dy, B.; McKenzie, T.; Thompson, G.; Wermers, R.; Lyden, M. Rates of hypercalcemia and hyperparathyroidism among patients with porcelain gallbladder. Am. J. Surg. 2020, 220, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Infection as a risk factor for gallbladder cancer. J. Surg. Oncol. 2006, 93, 633–639. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, B.Y.; Shi, J.S.; Wu, L.Q. Expression of the bacterial gene in gallbladder carcinoma tissue and bile. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 133–135. [Google Scholar] [PubMed]
- Iyer, P.; Barreto, S.G.; Sahoo, B.; Chandrani, P.; Ramadwar, M.R.; Shrikhande, S.V.; Dutt, A. Non-typhoidal Salmonella DNA traces in gallbladder cancer. Infect. Agents Cancer 2016, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Escobedo, G.; Marshall, J.M.; Gunn, J.S. Chronic and acute infection of the gall bladder by Salmonella Typhi: Understanding the carrier state. Nat. Rev. Microbiol. 2011, 9, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caygill, C.P.; Hill, M.J.; Braddick, M.; Sharp, J.C. Cancer mortality in chronic typhoid and paratyphoid carriers. Lancet 1994, 343, 83–84. [Google Scholar] [CrossRef]
- Shukla, V.K.; Singh, H.; Pandey, M.; Upadhyay, S.K.; Nath, G. Carcinoma of the gallbladder--is it a sequel of typhoid? Dig. Dis. Sci. 2000, 45, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Scanu, T.; Spaapen, R.M.; Bakker, J.M.; Pratap, C.B.; Wu, L.E.; Hofland, I.; Broeks, A.; Shukla, V.K.; Kumar, M.; Janssen, H.; et al. Salmonella Manipulation of Host Signaling Pathways Provokes Cellular Transformation Associated with Gallbladder Carcinoma. Cell Host Microbe 2015, 17, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Dewhirst, F.E.; Shen, Z.; Feng, Y.; Taylor, N.S.; Paster, B.J.; Ericson, R.L.; Lau, C.N.; Correa, P.; Araya, J.C.; et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 1998, 114, 755–763. [Google Scholar] [CrossRef]
- Matsukura, N.; Yokomuro, S.; Yamada, S.; Tajiri, T.; Sundo, T.; Hadama, T.; Kamiya, S.; Naito, Z.; Fox, J.G. Association between Helicobacter bilis in bile and biliary tract malignancies: H. bilis in bile from Japanese and Thai patients with benign and malignant diseases in the biliary tract. Jpn. J. Cancer Res. 2002, 93, 842–847. [Google Scholar] [CrossRef]
- Murata, H.; Tsuji, S.; Tsujii, M.; Fu, H.Y.; Tanimura, H.; Tsujimoto, M.; Matsuura, N.; Kawano, S.; Hori, M. Helicobacter bilis infection in biliary tract cancer. Aliment. Pharmacol. Ther. 2004, 20 (Suppl. 1), 90–94. [Google Scholar] [CrossRef]
- Prueksapanich, P.; Piyachaturawat, P.; Aumpansub, P.; Ridtitid, W.; Chaiteerakij, R.; Rerknimitr, R. Liver Fluke-Associated Biliary Tract Cancer. Gut Liver 2018, 12, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Zhu, Y.; Zhao, Z.; Wu, Z.; Okanurak, K.; Lv, Z. Liver fluke infection and cholangiocarcinoma: A review. Parasitol. Res. 2017, 116, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kamiza, A.B.; Su, F.H.; Wang, W.C.; Sung, F.C.; Chang, S.N.; Yeh, C.C. Chronic hepatitis infection is associated with extrahepatic cancer development: A nationwide population-based study in Taiwan. BMC Cancer 2016, 16, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsing, A.W.; Zhang, M.; Rashid, A.; McGlynn, K.A.; Wang, B.S.; Niwa, S.; Ortiz-Conde, B.A.; Goedert, J.J.; Fraumeni, J.F.; O’Brien, T.R.; et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: A population-based study in China. Int. J. Cancer 2008, 122, 1849–1853. [Google Scholar] [CrossRef]
- Choe, J.W.; Hyun, J.J.; Kim, B.; Han, K.D. Influence of Metabolic Syndrome on Cancer Risk in HBV Carriers: A Nationwide Population Based Study Using the National Health Insurance Service Database. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.A. Early diagnosis of fungal infection in immunocompromised patients. J. Antimicrob. Chemother. 2008, 61 (Suppl. S1), i3–i6. [Google Scholar] [CrossRef]
- Szvalb, A.D.; Kontoyiannis, D.P. Acute acalculous cholecystitis due to Fusarium species and review of the literature on fungal cholecystitis. Mycoses 2019, 62, 847–853. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Giat, E.; Ehrenfeld, M.; Shoenfeld, Y. Cancer and autoimmune diseases. Autoimmun. Rev. 2017, 16, 1049–1057. [Google Scholar] [CrossRef]
- Castro, F.A.; Liu, X.; Försti, A.; Ji, J.; Sundquist, J.; Sundquist, K.; Koshiol, J.; Hemminki, K. Increased risk of hepatobiliary cancers after hospitalization for autoimmune disease. Clin. Gastroenterol. Hepatol. 2014, 12, 1038–1045. [Google Scholar] [CrossRef]
- Horsley-Silva, J.L.; Rodriguez, E.A.; Franco, D.L.; Lindor, K.D. An update on cancer risk and surveillance in primary sclerosing cholangitis. Liver Int. 2017, 37, 1103–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burak, K.; Angulo, P.; Pasha, T.M.; Egan, K.; Petz, J.; Lindor, K.D. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am. J. Gastroenterol. 2004, 99, 523–526. [Google Scholar] [CrossRef] [PubMed]
- de Valle, M.B.; Björnsson, E.; Lindkvist, B. Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort. Liver Int. 2012, 32, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013, 58, 2045–2055. [Google Scholar] [CrossRef]
- Barner-Rasmussen, N.; Pukkala, E.; Jussila, A.; Färkkilä, M. Epidemiology, risk of malignancy and patient survival in primary sclerosing cholangitis: A population-based study in Finland. Scand. J. Gastroenterol. 2020, 55, 74–81. [Google Scholar] [CrossRef]
- Lewis, J.T.; Talwalkar, J.A.; Rosen, C.B.; Smyrk, T.C.; Abraham, S.C. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: Evidence for a metaplasia-dysplasia-carcinoma sequence. Am. J. Surg. Pathol. 2007, 31, 907–913. [Google Scholar] [CrossRef]
- Buckles, D.C.; Lindor, K.D.; Larusso, N.F.; Petrovic, L.M.; Gores, G.J. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am. J. Gastroenterol. 2002, 97, 1138–1142. [Google Scholar] [CrossRef]
- Said, K.; Glaumann, H.; Björnstedt, M.; Bergquist, A. The value of thioredoxin family proteins and proliferation markers in dysplastic and malignant gallbladders in patients with primary sclerosing cholangitis. Dig. Dis. Sci. 2012, 57, 1163–1170. [Google Scholar] [CrossRef]
- Wilsnack, R.W.; Wilsnack, S.C.; Kristjanson, A.F.; Vogeltanz-Holm, N.D.; Gmel, G. Gender and alcohol consumption: Patterns from the multinational GENACIS project. Addiction 2009, 104, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Neighbors, C.; Gilson, M.; Larimer, M.E.; Alan Marlatt, G. Epidemiological trends in drinking by age and gender: Providing normative feedback to adults. Addict. Behav. 2007, 32, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Delker, E.; Brown, Q.; Hasin, D.S. Alcohol Consumption in Demographic Subpopulations: An Epidemiologic Overview. Alcohol Res. 2016, 38, 7–15. [Google Scholar] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef]
- Acalovschi, M. Gallstones in patients with liver cirrhosis: Incidence, etiology, clinical and therapeutical aspects. World J. Gastroenterol. 2014, 20, 7277–7285. [Google Scholar] [CrossRef]
- Del Olmo, J.A.; García, F.; Serra, M.A.; Maldonado, L.; Rodrigo, J.M. Prevalence and incidence of gallstones in liver cirrhosis. Scand. J. Gastroenterol. 1997, 32, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M. Analytical methods for aflatoxins in corn and peanuts. Arch. Environ. Contam. Toxicol. 1989, 18, 308–314. [Google Scholar] [CrossRef]
- Scholl, P.F.; Groopman, J.D. Long-term stability of human aflatoxin B1 albumin adducts assessed by isotope dilution mass spectrometry and high-performance liquid chromatography-fluorescence. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1436–1439. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, C.; Koshiol, J.; Guerrero, A.R.; Kogan, M.J.; Ferreccio, C. The case for aflatoxins in the causal chain of gallbladder cancer. Med. Hypotheses 2016, 86, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Emerole, G.O. Excretion of aflatoxin B1 as a glutathione conjugate. Eur. J. Drug Metab. Pharm. 1981, 6, 265–268. [Google Scholar] [CrossRef]
- Harland, E.C.; Cardeilhac, P.T. Excretion of carbon-14-labeled aflatoxin B1 via bile, urine, and intestinal contents of the chicken. Am. J. Vet. Res. 1975, 36, 909–912. [Google Scholar] [PubMed]
- Nogueira, L.; Foerster, C.; Groopman, J.; Egner, P.; Koshiol, J.; Ferreccio, C.; Group, G.C.C.W. Association of aflatoxin with gallbladder cancer in Chile. JAMA 2015, 313, 2075–2077. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Gao, Y.T.; Dean, M.; Egner, P.; Nepal, C.; Jones, K.; Wang, B.; Rashid, A.; Luo, W.; Van Dyke, A.L.; et al. Association of Aflatoxin and Gallbladder Cancer. Gastroenterology 2017, 153, 488–494.e481. [Google Scholar] [CrossRef] [PubMed]
- Raju, N.J. Arsenic in the geo-environment: A review of sources, geochemical processes, toxicity and removal technologies. Environ. Res. 2021, 203, 111782. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011, 2011, 431287. [Google Scholar] [CrossRef] [Green Version]
- Tapio, S.; Grosche, B. Arsenic in the aetiology of cancer. Mutat. Res. 2006, 612, 215–246. [Google Scholar] [CrossRef]
- Ganesan, N.; Bambino, K.; Boffetta, P.; Labgaa, I. Exploring the potential carcinogenic role of arsenic in gallbladder cancer. Eur. J. Cancer Prev. 2020, 29, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Gao, Y.T.; Huang, Y.H.; McGee, E.E.; Lam, T.; Wang, B.; Shen, M.C.; Rashid, A.; Pfeiffer, R.M.; Hsing, A.W.; et al. A Metallomic Approach to Assess Associations of Serum Metal Levels With Gallstones and Gallbladder Cancer. Hepatology 2020, 71, 917–928. [Google Scholar] [CrossRef]
- Pandey, M. Environmental pollutants in gallbladder carcinogenesis. J. Surg. Oncol. 2006, 93, 640–643. [Google Scholar] [CrossRef]
- Versieck, J.; Hoste, J.; Vanballenberghe, L.; Barbier, F.; Cornelis, R.; Waelput, I. Serum molybdenum in diseases of the liver and biliary system. J. Lab. Clin. Med. 1981, 97, 535–544. [Google Scholar] [PubMed]
- Shukla, V.K.; Adukia, T.K.; Singh, S.P.; Mishra, C.P.; Mishra, R.N. Micronutrients, antioxidants, and carcinoma of the gallbladder. J. Surg. Oncol. 2003, 84, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Sreenivas, V.; Velpandian, T.; Kapil, U.; Garg, P.K. Risk factors for gallbladder cancer: A case-control study. Int. J. Cancer 2013, 132, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Tomásek, L.; Darby, S.C.; Swerdlow, A.J.; Placek, V.; Kunz, E. Radon exposure and cancers other than lung cancer among uranium miners in West Bohemia. Lancet 1993, 341, 919–923. [Google Scholar] [CrossRef]
- Gröber, U.; Holzhauer, P.; Kisters, K.; Holick, M.F.; Adamietz, I.A. Micronutrients in Oncological Intervention. Nutrients 2016, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.A.; Marcano-Bonilla, L.; Roberts, L.R. Gallbladder cancer: Epidemiology and genetic risk associations. Chin. Clin. Oncol. 2019, 8, 31. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Thandra, K.C.; Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 2019, 5, 93–102. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.Y.; Zhu, A.X. Biliary tract cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef]
- Aljindan, R.Y.; Alkharsah, K.R. Pattern of increased antimicrobial resistance of Salmonella isolates in the Eastern Province of KSA. J. Taibah Univ. Med. Sci. 2020, 15, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Medalla, F.; Gu, W.; Friedman, C.R.; Judd, M.; Folster, J.; Griffin, P.M.; Hoekstra, R.M. Increased Incidence of Antimicrobial-Resistant Nontyphoidal Salmonella Infections, United States, 2004-2016. Emerg. Infect. Dis. 2021, 27, 1662–1672. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Moreno, P.; Riquelme, I.; García, P.; Brebi, P.; Roa, J.C. Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer. J. Pers. Med. 2022, 12, 234. https://doi.org/10.3390/jpm12020234
Pérez-Moreno P, Riquelme I, García P, Brebi P, Roa JC. Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer. Journal of Personalized Medicine. 2022; 12(2):234. https://doi.org/10.3390/jpm12020234
Chicago/Turabian StylePérez-Moreno, Pablo, Ismael Riquelme, Patricia García, Priscilla Brebi, and Juan Carlos Roa. 2022. "Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer" Journal of Personalized Medicine 12, no. 2: 234. https://doi.org/10.3390/jpm12020234
APA StylePérez-Moreno, P., Riquelme, I., García, P., Brebi, P., & Roa, J. C. (2022). Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer. Journal of Personalized Medicine, 12(2), 234. https://doi.org/10.3390/jpm12020234