
Evolution of Protein Functional Annotation: Text Mining Study

 , ,
, ,
Abstract
:
1. Introduction
- Sequence and structural analysis;
- Subcellular localization(s);
- Protein–protein interactions (Guilty by Association);
- Expression and coexpression;
- Phenotypes and diseases;
- Text mining (including GO annotation).
2. Materials and Methods
2.1. neXtProt Versions
2.2. Instruments to Compare neXtProt Versions
- (1)
- heatmap: Pretty Heatmaps, Raivo Kolde, 2019, R package version 1.0.12, https://CRAN.R-project.org/package=pheatmap (accessed on 11 March 2022);
- (2)
- wordcloud2: Create Word Cloud by “htmlwidget”, Dawei Lang and Guan-tin Chien, 2018, R package version 0.2.1, https://CRAN.R-project.org/package=wordcloud2 (accessed on 11 March 2022);
- (3)
- viridis: Default Color Maps from “matplotlib”, Simon Garnier, 2018, R package version 0.5.1, https://CRAN.R-project.org/package=viridis (accessed on 11 March 2022);
- (4)
- viridisLite: Default Color Maps from “matplotlib” (Lite Version), Simon Garnier, 2018, R package version 0.3.0, https://CRAN.R-project.org/package=viridisLite (accessed on 11 March 2022);
- (5)
- gridExtra: Miscellaneous Functions for “Grid” Graphics, Baptiste Auguie, 2017, R package version 2.3, https://CRAN.R-project.org/package=gridExtra (accessed on 11 March 2022);
- (6)
- RColorBrewer: ColorBrewer Palettes, Erich Neuwirth, 2014, R package version 1.1-2, https://CRAN.R-project.org/package=RColorBrewer (accessed on 11 March 2022);
- (7)
- forcats: Tools for Working with Categorical Variables (Factors), Hadley Wickham, 2020, R package version 0.5.0, https://CRAN.R-project.org/package=forcats (accessed on 11 March 2022);
- (8)
- stringr: Simple, Consistent Wrappers for Common String Operations, Hadley Wickham, 2019, R;
- (9)
- dplyr: A Grammar of Data Manipulation, Hadley Wickham and Romain Francois and Lionel Henry and Kirill Muller, 2021, R package version 1.0.7, https://CRAN.R-project.org/package=dplyr (accessed on 11 March 2022);
- (10)
- purrr: Functional Programming Tools, Lionel Henry and Hadley Wickham, 2020, R package version 0.3.4, https://CRAN.R-project.org/package=purrr (accessed on 11 March 2022).
3. Results
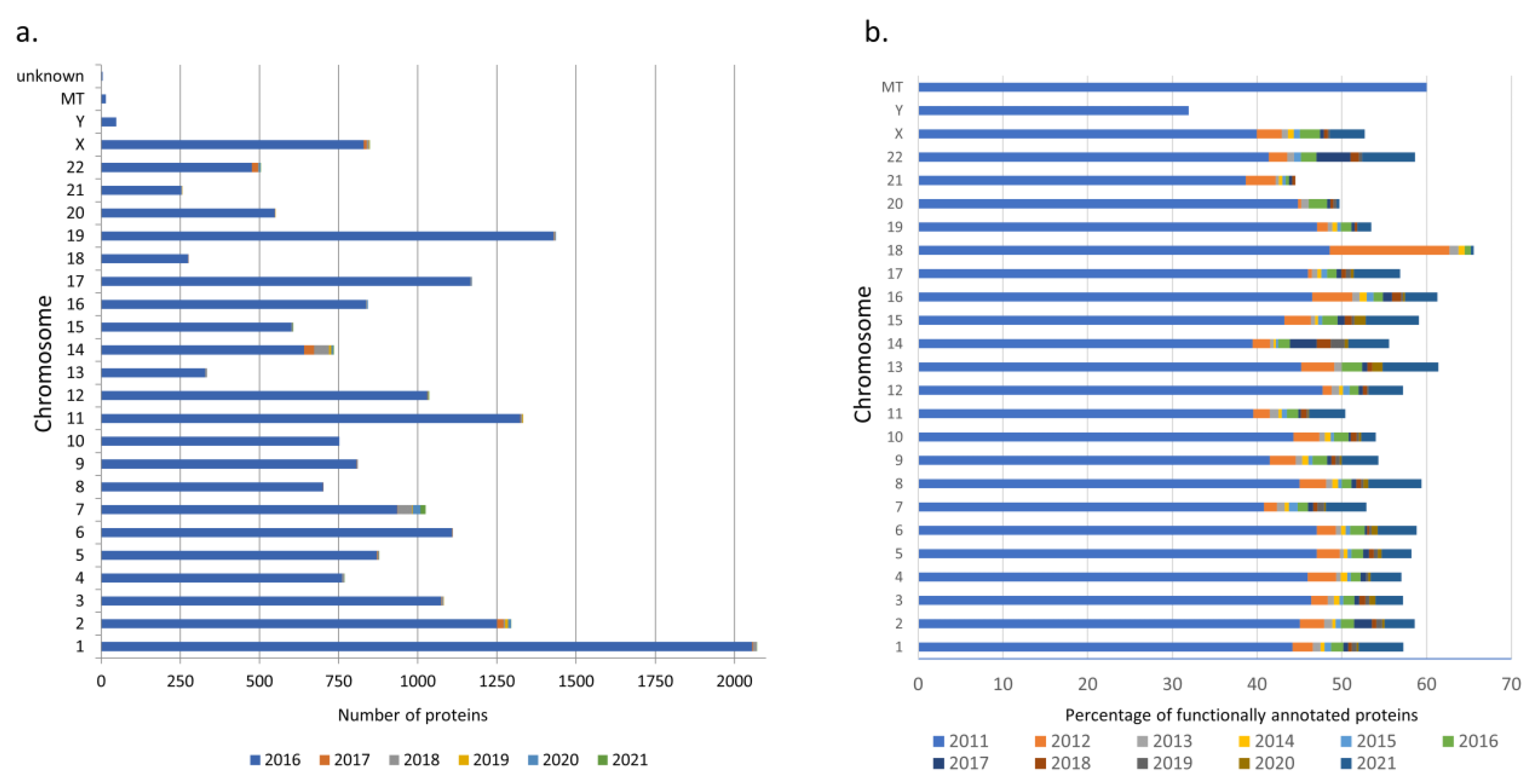
3.1. neXtProt Version Comparison
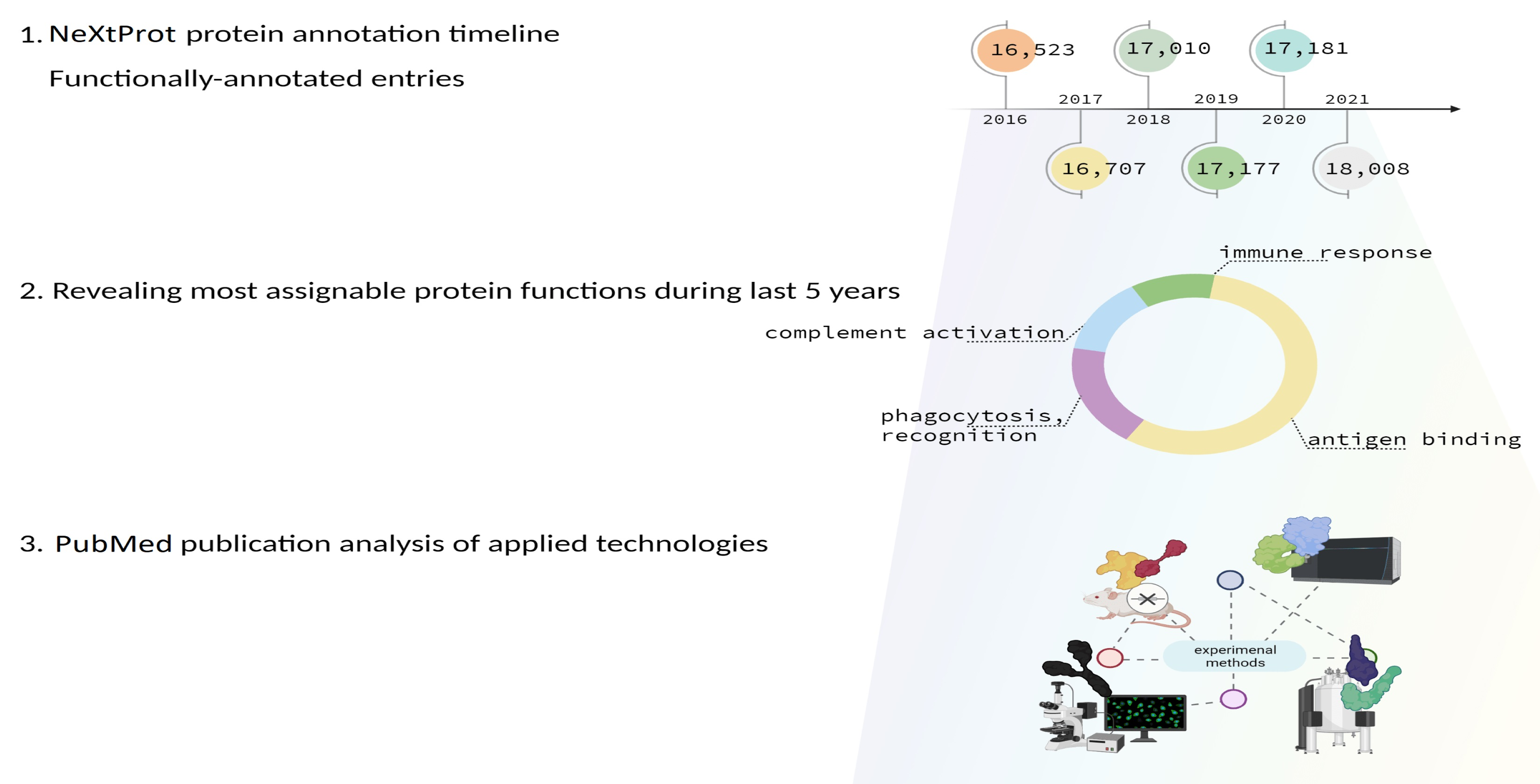
3.2. Proteins Whose Functions Have Been Discovered over the Past Five Years According to Data on neXtProt
3.3. Analysis of Publications Describing Experiments during Which the Functions of Proteins Were First Established in the Period from 2016 to 2021
- On average, a publication confirms the functions of 3.81 proteins.
- In 135 publications, data are provided on the function of only one (but not the same, of course) protein.
- On average, there are 1.5 publications per protein with confirmation of its functions.
- Overall, 611 proteins have a confirmation of function in only one publication.
- In all, 58% of all the functions shown were binding to other proteins (“protein binding”).
- The publication dates for the candidate articles under consideration were extracted from neXtProt and, if necessary, supplemented (in some cases, only the year was indicated for publication, which may not be enough to determine the primacy).
- The list of publications, evidence, annotations, and proteins was sorted by publication date and protein identifier.
- Multiple records of protein function were deleted, except for the one relying on the oldest publication for the period under review.
- In this way, records were deleted regarding all the functions, except for one. Several protein functions could be determined; the missing data were then reattached by the coincidence of the identifiers of the protein and publication.
- AP-MS: affinity purification of labeled (3xFLAG) and interacting proteins with subsequent mass spectrometric identification of the components of the complex.
- LUMIER: a method based on copurification and luminescence of complexes: the gene of one of the potential participants of the complex was fused with a sequence encoding luciferase and stably expressed in 293T cell lines; genes of other potential partners with 3xFLAG incorporation were transfected into cells of the reporter line. Cell lysates were incubated together in the presence of antibodies for the label (3xFLAG), and after washing, the formation of a stable complex was determined by the presence of luminescence.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Auton, A.; Brooks, L.; Durbin, R.; Garrison, E.; Kang, H.; Korbel, J.; Marchini, J.; McCarthy, S.; McVean, G.; Abecasis, G. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.; Minikel, K.; Samocha, K.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.; Ware, J.; Hill, A.; Cummings, B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.-K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A High-Stringency Blueprint of the Human Proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Cristea, I.M.; Corrales, F.J.; Lindskog, C.; Paik, Y.-K.; Van Eyk, J.E.; Liu, S.; Pennington, S.R.; et al. Research on the Human Proteome Reaches a Major Milestone: >90% of Predicted Human Proteins Now Credibly Detected, According to the HUPO Human Proteome Project. J. Proteome Res. 2020, 19, 4735. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S. Reflections on the HUPO Human Proteome Project, the Flagship Project of the Human Proteome Organization, at 10 Years. Mol. Cell. Proteom. 2021, 20, 100062. [Google Scholar] [CrossRef]
- Rembeza, E.; Engqvist, M.K.M. Experimental and Computational Investigation of Enzyme Functional Annotations Uncovers Misannotation in the EC 1.1.3.15 Enzyme Class. PLoS Comput. Biol. 2021, 17, e1009446. [Google Scholar] [CrossRef]
- Bruey, J.M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL Regulate Proinflammatory Caspase-1 Activation by Interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Duek, P.; Mary, C.; Zahn-Zabal, M.; Bairoch, A.; Lane, L. Functionathon: A Manual Data Mining Workflow to Generate Functional Hypotheses for Uncharacterized Human Proteins and Its Application by Undergraduate Students. Database 2021, 2021, baab046. [Google Scholar] [CrossRef]
- Rost, B.; Liu, J. The PredictProtein Server. Nucleic Acids Res. 2003, 31, 3300–3304. [Google Scholar] [CrossRef]
- Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 11 March 2022).
- The Gene Ontology Consortium the Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [Green Version]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How Many Human Proteoforms Are There? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Bludau, I.; Frank, M.; Dörig, C.; Cai, Y.; Heusel, M.; Rosenberger, G.; Picotti, P.; Collins, B.C.; Röst, H.; Aebersold, R. Systematic Detection of Functional Proteoform Groups from Bottom-up Proteomic Datasets. Nat. Commun. 2021, 12, 3810. [Google Scholar] [CrossRef]
- Poverennaya, E.; Kiseleva, O.; Romanova, A.; Pyatnitskiy, M. Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms. Genes 2020, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Zahn-Zabal, M.; Michel, P.A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; De Laval, V.R.; et al. The NeXtProt Knowledgebase in 2020: Data, Tools and Usability Improvements. Nucleic Acids Res. 2020, 48, D328–D334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duek, P.; Gateau, A.; Bairoch, A.; Lane, L. Exploring the Uncharacterized Human Proteome Using NeXtProt. J. Proteome Res. 2018, 17, 4211–4226. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.K.; Lane, L.; Kawamura, T.; Chen, Y.J.; Cho, J.Y.; Labaer, J.; Yoo, J.S.; Domont, G.; Corrales, F.; Omenn, G.S.; et al. Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res. 2018, 17, 4042–4050. [Google Scholar] [CrossRef]
- Sael, L.; Chitale, M.; Kihara, D. Structure- and Sequence-Based Function Prediction for Non- Homologous Proteins. J. Struct. Funct. Genom. 2012, 13, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Kulmanov, M.; Hoehndorf, R. DeepGOPlus: Improved Protein Function Prediction from Sequence. Bioinformatics 2020, 36, 422–429. [Google Scholar] [CrossRef]
- You, R.; Yao, S.; Mamitsuka, H.; Zhu, S. DeepGraphGO: Graph Neural Network for Large-Scale, Multispecies Protein Function Prediction. Bioinformatics 2021, 37, I262–I271. [Google Scholar] [CrossRef]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-Throughput Functional Annotation of Uncharacterized Proteins in an Error-Prone Environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; You, R.; Wang, S.; Xiong, Y.; Huang, X.; Zhu, S. NetGO 2.0: Improving Large-Scale Protein Function Prediction with Massive Sequence, Text, Domain, Family and Network Information. Nucleic Acids Res. 2021, 49, W469–W475. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Berriman, M.; Barton, G. GOtcha: A New Method for Prediction of Protein Function Assessed by the Annotation of Seven Genomes. BMC Bioinform. 2004, 5, 178. [Google Scholar] [CrossRef] [Green Version]
- Falda, M.; Toppo, S.; Pescarolo, A.; Lavezzo, E.; Di Camillo, B.; Facchinetti, A.; Cilia, E.; Velasco, R.; Fontana, P. Argot2: A Large Scale Function Prediction Tool Relying on Semantic Similarity of Weighted Gene Ontology Terms. BMC Bioinform. 2012, 13 (Suppl. 4), S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, S.; García-Gómez, J.; Terol, J.; Williams, T.; Nagaraj, S.; Nueda, M.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-Throughput Functional Annotation and Data Mining with the Blast2GO Suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Zhang, C.; Lane, L.; Omenn, G.S.; Zhang, Y. Blinded Testing of Function Annotation for UPE1 Proteins by I-TASSER/COFACTOR Pipeline Using the 2018–2019 Additions to NeXtProt and the CAFA3 Challenge. J. Proteome Res. 2019, 18, 4154. [Google Scholar] [CrossRef]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, M.N.; et al. The CAFA Challenge Reports Improved Protein Function Prediction and New Functional Annotations for Hundreds of Genes through Experimental Screens. Genome Biol. 2019, 20, 244. [Google Scholar] [CrossRef] [Green Version]
- Rolland, T.; Ta\csan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A Proteome-Scale Map of the Human Interactome Network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.; Larsen, B.; Choi, H. A Quantitative Chaperone Interaction Network Reveals the Architecture of Cellular Protein Homeostasis Pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, A.; Benlasfer, N.; Birth, P.; Hegele, A.; Wachsmuth, F.; Apelt, L.; Stelzl, U. Phospho-Tyrosine Dependent Protein—Protein Interaction Network. Mol. Syst. Biol. 2015, 11, 794. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Overall, C.M.; Van Eyk, J.E.; Baker, M.S.; Paik, Y.K.; Weintraub, S.T.; Lane, L.; Martens, L.; Vandenbrouck, Y.; Kusebauch, U.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Lane, L.; Overall, C.M.; Bandeira, N.; Baker, M.S.; Pineau, C.; Moritz, R.L.; Corrales, F.; Orchard, S.; Van Eyk, J.E.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res. 2019, 18, 4108–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A Draft Map of the Human Proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Savitski, M.M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteom. MCP 2015, 14, 2394. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Farrah, T.; Lam, H.; Tasman, N.; Sun, Z.; Nilsson, E.; Pratt, B.; Prazen, B.; et al. A Guided Tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10, 1150. [Google Scholar] [CrossRef] [Green Version]
- Zahn-Zabal, M.; Lane, L. What Will NeXtProt Help Us Achieve in 2020 and Beyond? Expert Rev. Proteom. 2020, 17, 95–98. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Categories | Number of Records |
|---|---|
| catalytic-activity | 39 |
| function-info | 802 |
| go-biological-process | 2684 |
| go-molecular-function | 1571 |
| Pathway | 1303 |
| transport-activity | 98 |
| Functions Detected More Frequently | Functions Detected Less Frequently |
|---|---|
| antigen binding | ATP binding |
| immunoglobulin production | DNA-binding transcription factor activity, RNA polymerase II-specific |
| immunoglobulin receptor binding | DNA binding |
| phagocytosis, recognition | DNA-binding transcription factor activity |
| positive regulation of B cell activation | regulation of transcription by RNA polymerase II |
| phagocytosis, engulfment | positive regulation of transcription, DNA-templated |
| complement activation, classical pathway | oxidation–reduction process |
| B cell receptor signaling pathway | positive regulation of transcription by RNA polymerase II |
| immune response | protein serine/threonine kinase activity |
| defense response to bacterium | regulation of transcription, DNA-templated |
| adaptive immune response |
| The Sequence of Identification of Protein Functions | Number of Cases |
|---|---|
| Protein binding and any other function have been identified in one publication | 79 |
| Protein binding has been shown prior to determining any other function | 58 |
| Protein binding has been shown after determining any other function | 14 |
| Protein binding has not been shown, but other function has been defined | 73 |
| To date, only protein binding has been shown | 442 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilgisonis, E.V.; Pogodin, P.V.; Kiseleva, O.I.; Tarbeeva, S.N.; Ponomarenko, E.A. Evolution of Protein Functional Annotation: Text Mining Study. J. Pers. Med. 2022, 12, 479. https://doi.org/10.3390/jpm12030479
Ilgisonis EV, Pogodin PV, Kiseleva OI, Tarbeeva SN, Ponomarenko EA. Evolution of Protein Functional Annotation: Text Mining Study. Journal of Personalized Medicine. 2022; 12(3):479. https://doi.org/10.3390/jpm12030479
Chicago/Turabian StyleIlgisonis, Ekaterina V., Pavel V. Pogodin, Olga I. Kiseleva, Svetlana N. Tarbeeva, and Elena A. Ponomarenko. 2022. "Evolution of Protein Functional Annotation: Text Mining Study" Journal of Personalized Medicine 12, no. 3: 479. https://doi.org/10.3390/jpm12030479
APA StyleIlgisonis, E. V., Pogodin, P. V., Kiseleva, O. I., Tarbeeva, S. N., & Ponomarenko, E. A. (2022). Evolution of Protein Functional Annotation: Text Mining Study. Journal of Personalized Medicine, 12(3), 479. https://doi.org/10.3390/jpm12030479