
Physiologically-Based Pharmacokinetics Modeling for Hydroxychloroquine as a Treatment for Malaria and Optimized Dosing Regimens for Different Populations



Abstract
:1. Introduction
2. Method
2.1. The PBPK Model Construction and Verification for HCQ
2.2. The Inclusion of Different Populations
3. Results
3.1. The PBPK Model for HCQ
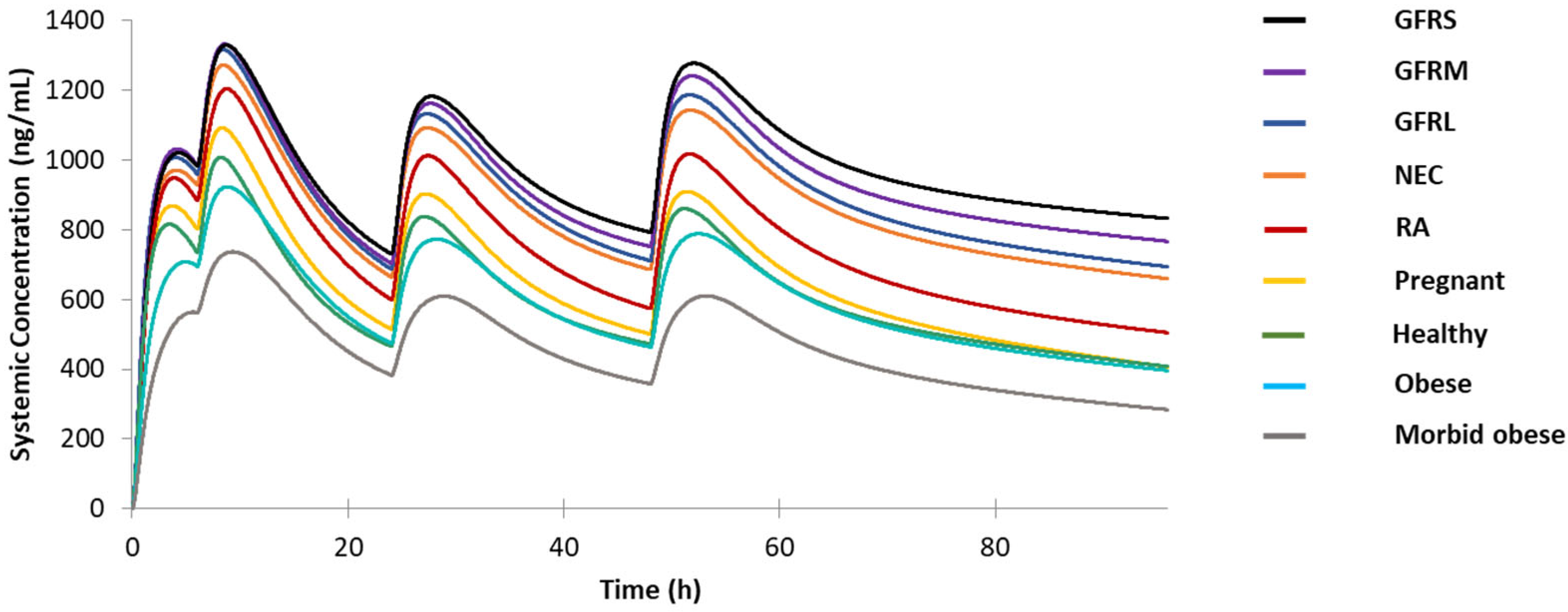
3.2. Simulation on the Original Dosage Regimen for Different Special Populations
3.3. Dosing Adjustment Recommendation for Different Populations
3.3.1. Pregnancy Population
3.3.2. RA Population
3.3.3. NEC Population
3.3.4. Renal Impairment Populations
3.3.5. Obese Population and MO Population
3.3.6. Pediatric Population
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Malaria, R.B. World Malaria Report 2005; World Health Organization: Geneva, Switzerland; UNICEF: New York, NY, USA, 2005.
- World Health Organization. World Malaria Report 2015; World Health Organization: Geneva, Switzerland, 2016.
- White, N.J. The treatment of malaria. N. Engl. J. Med. 1996, 335, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Good, M.F.; Milon, G. Malaria Pathogenesis. Science 1994, 264, 1878–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangpukdee, N.; Duangdee, C.; Wilairatana, P.; Krudsood, S. Malaria diagnosis: A brief review. Korean J. Parasitol. 2009, 47, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/009768s037s045s047lbl.pdf (accessed on 18 November 2021).
- Wellems, T.E.; Plowe, C.V. Chloroquine-Resistant Malaria. J. Infect. Dis. 2001, 184, 770–776. [Google Scholar] [CrossRef]
- Sahraei, Z.; Shabani, M.; Shokouhi, S.; Saffaei, A. Aminoquinolines against coronavirus disease 2019 (COVID-19): Chloroquine or hydroxychloroquine. Int. J. Antimicrob. Agents 2020, 55, 105945. [Google Scholar] [CrossRef]
- Klinger, G.; Morad, Y.; Westall, C.A.; Laskin, C.; Spitzer, K.A.; Koren, G.; Ito, S.; Buncic, R.J. Ocular toxicity and antenatal exposure to chloroquine or hydroxychloroquine for rheumatic diseases. Lancet 2001, 358, 813–814. [Google Scholar] [CrossRef]
- Motta, M.; Tincani, A.; Faden, D.; Zinzini, E.; Lojacono, A.; Marchesi, A.; Frassi, M.; Biasini, C.; Zatti, S.; Chirico, G. Follow-up of infants exposed to hydroxychloroquine given to mothers during pregnancy and lactation. J. Perinatol. 2005, 25, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef]
- Furst, D.E. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996, 5, 11–15. [Google Scholar] [CrossRef]
- Colson, P.; Rolain, J.-M.; Lagier, J.-C.; Brouqui, P.; Raoult, D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105932. [Google Scholar] [CrossRef]
- Marquardt, K.; Albertson, T.E. Treatment of hydroxychloroquine overdose. Am. J. Emerg. Med. 2001, 19, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Doyno, C.; Sobieraj, D.M.; Baker, W.L. Toxicity of chloroquine and hydroxychloroquine following therapeutic use or overdose. Clin. Toxicol. 2021, 59, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Long, N. Chloroquine and Hydroxychloroquine. Toxicology 2020. Available online: https://litfl.com/chloroquine-hydroxychloroquine-toxicity/ (accessed on 18 November 2021).
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: A systematic review of published models, applications, and model verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- Njaria, P.M.; Okombo, J.; Njuguna, N.M.; Chibale, K. Chloroquine-containing compounds: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1003–1024. [Google Scholar] [CrossRef]
- Romanelli, F.; Smith, K.M.; Hoven, A.D. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr. Pharm. Des. 2004, 10, 2643–2648. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp® Population-based ADME Simulator. Expert Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef]
- Hassan, S.F.; Rashid, U.; Ansari, F.L.; Ul-Haq, Z. Bioisosteric approach in designing new monastrol derivatives: An investigation on their ADMET prediction using in silico derived parameters. J. Mol. Graph. Model. 2013, 45, 202–210. [Google Scholar] [CrossRef]
- Hecht, D.; Fogel, G.B. Computational intelligence methods for ADMET prediction. Front. Drug Des. Discov. 2009, 4, 351–377. [Google Scholar]
- Gobeau, N.; Stringer, R.; De Buck, S.; Tuntland, T.; Faller, B. Evaluation of the GastroPlus™ advanced compartmental and transit (acat) model in early discovery. Pharm. Res. 2016, 33, 2126–2139. [Google Scholar] [CrossRef]
- Rayat, C.S. Statistical Methods in Medical Research; Springer: Berlin/Heidelberg, Germany, 2018; pp. 139–146. [Google Scholar]
- Zhai, J.; Ji, B.; Liu, S.; Zhang, Y.; Cai, L.; Wang, J. In Silico Prediction of Pharmacokinetic Profile for Human Oral Drug Candidates Which Lack Clinical Pharmacokinetic Experiment Data. Eur. J. Drug Metab. Pharmacokinet. 2022, 47, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, H.; Hjorton, K.; Abujrais, S.; Rönnblom, L.; Åkerfeldt, T.; Kultima, K. Measurement of hydroxychloroquine in blood from SLE patients using LC-HRMS—Evaluation of whole blood, plasma, and serum as sample matrices. Arthritis Res. Ther. 2020, 22, 125. [Google Scholar] [CrossRef] [PubMed]
- Tett, S.; Cutler, D.; Day, R.; Brown, K. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br. J. Clin. Pharmacol. 1989, 27, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tett, S.; Cutler, D.; Day, R.; Brown, K. A dose-ranging study of the pharmacokinetics of hydroxy-chloroquine following intravenous administration to healthy volunteers. Br. J. Clin. Pharmacol. 1988, 26, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Vinayagamoorthy, N.; Han, K.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Jung, S.-H.; Park, S.-W.; Chung, Y.-J. Association of polymorphisms of cytochrome P450 2D6 with blood hydroxychloroquine levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2016, 68, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quraishi, S.; Reed, B.T.; Duvoisin, R.M.; Taylor, W.R. Selective activation of mGluR8 receptors modulates retinal ganglion cell light responses. Neuroscience 2021, 166, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Crowe, A.; Ilett, K.F.; Karunajeewa, H.A.; Batty, K.T.; Davis, T.M.E. Role of P Glycoprotein in Absorption of Novel Antimalarial Drugs. Antimicrob. Agents Chemother. 2006, 50, 3504. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhu, L.; Chan, T.; Lu, X.; Shen, W.; Madigan, M.C.; Gillies, M.C.; Zhou, F. Chloroquine and Hydroxychloroquine Are Novel Inhibitors of Human Organic Anion Transporting Polypeptide 1A2. J. Pharm. Sci. 2016, 105, 884–890. [Google Scholar] [CrossRef]
- Collins, K.P.; Jackson, K.M.; Gustafson, D.L. Hydroxychloroquine: A physiologically-based pharmacokinetic model in the context of cancer-related autophagy modulation. J. Pharmacol. Exp. Ther. 2018, 365, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.M. Hydroxychloroquine Dimers as Inhibitors of Plasmodium Falciparum Chloroquine Resistant Transporter and P-glycoprotein. Master’s Thesis, Purdue University, West Lafayette, IN, USA, 2015. [Google Scholar]
- Tailor, R.; Elaraoud, I.; Good, P.; Hope-Ross, M.; Scott, R.A.H. A Case of Severe Hydroxychloroquine-Induced Retinal Toxicity in a Patient with Recent Onset of Renal Impairment: A Review of the Literature on the Use of Hydroxychloroquine in Renal Impairment. Case Rep. Ophthalmol. Med. 2012, 2012, 182747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Huang, Y.; Ye, Q.; Shang, F.; Ming, M.; Xu, H.; Li, Z. Low-dose oral hydroxychloroquine led to impaired vision in a child with renal failure: Case report and literature review. Medicine 2021, 100, e24919. [Google Scholar] [CrossRef] [PubMed]
- El-Solia, A.; Al-Otaibi, K.; Ai-Hwiesh, A.K. Hydroxychloroquine-induced hypoglycaemia in non-diabetic renal patient on peritoneal dialysis. Case Rep. 2018, 2018, bcr-2017-223639. [Google Scholar] [CrossRef] [PubMed]
- Dogar, M.U.; Shah, N.; Ishtiaq, S.; Shah, P.N.; Shah, P.; Mathew, S.; Vittorio, T.J. Hydroxychloroquine-induced restrictive cardiomyopathy: A case report. Postgrad. Med. J. 2018, 94, 185–186. [Google Scholar] [CrossRef] [PubMed]
- O’Laughlin, J.P.; Mehta, P.H.; Wong, B.C. Life threatening severe QTc prolongation in patient with systemic lupus erythematosus due to hydroxychloroquine. Case Rep. Cardiol. 2016, 2016, 4626279. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, T.; Kupa, L.D.V.K.; Pasoto, S.G.; Aikawa, N.E.; Borba, E.F.; Duarte, N.J.; Leon, E.P.; Silva, C.A.; Bonfá, E. The influence of obesity on hydroxychloroquine blood levels in lupus nephritis patients. Lupus 2021, 30, 554–559. [Google Scholar] [CrossRef]
- Sitar, D.S. Aging issues in drug disposition and efficacy. Proc. West Pharmacol. Soc. 2007, 50, 16–20. [Google Scholar]
- Stewart, C.; Hampton, E. Effect of maturation on drug disposition in pediatric patients. Clin. Pharm. 1987, 6, 548–564. [Google Scholar]
- Van Der Heijde, D.; Van Riel, P.; Gribnau, F.; Nuver-Zwart, I.; Van De Putte, L. Effects of hydroxychloroquine and sulphasalazine on progression of joint damage in rheumatoid arthritis. Lancet 1989, 333, 1036–1038. [Google Scholar] [CrossRef]
- Lim, H.-S.; Im, J.-S.; Cho, J.-Y.; Bae, K.-S.; Klein, T.A.; Yeom, J.-S.; Kim, T.-S.; Choi, J.-S.; Jang, I.-J.; Park, J.-W. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob. Agents Chemother. 2009, 53, 1468–1475. [Google Scholar] [CrossRef] [Green Version]
- Tsujimura, S.; Saito, K.; Nawata, M.; Nakayamada, S.; Tanaka, Y. Overcoming drug resistance induced by P-glycoprotein on lymphocytes in patients with refractory rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 380–388. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrisette, T.; Lodise, T.P.; Scheetz, M.H.; Goswami, S.; Pogue, J.M.; Rybak, M.J. The pharmacokinetic and pharmacodynamic properties of hydroxychloroquine and dose selection for COVID-19: Putting the cart before the horse. Infect. Dis. Ther. 2020, 9, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Zhang, X.; Liu, J.; Yang, Y.; Zheng, N.; Liu, Q.; Bergman, K.; Reynolds, K.; Huang, S.-M.; Zhu, H.; et al. Connecting Hydroxychloroquine In Vitro Antiviral Activity to In Vivo Concentration for Prediction of Antiviral Effect: A Critical Step in Treating Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 3232–3236. [Google Scholar] [CrossRef] [PubMed]
- Yeo, K.R.; Zhang, M.; Pan, X.; Ke, A.B.; Jones, H.M.; Wesche, D.; Almond, L.M. Impact of disease on plasma and lung exposure of chloroquine, hydroxychloroquine and azithromycin: Application of PBPK modeling. Clin. Pharmacol. Ther. 2020, 108, 976–984. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Input Value |
|---|---|
| Physiochemical Properties | |
| Molecular weight (g/mol) | 335.872 a |
| LogP | 3.6 b |
| pKa | 9.67 b |
| Blood Binding | |
| B/P | 0.55 d |
| Fu | 0.1 d |
| Absorption (ADAM model) | |
| Peff (10−4 cm/s) | 2.32 c |
| PSA (Å2) | 48.39 a |
| Distribution (Full PBPK model) | |
| Vss (L/kg) | SimCYP predicted |
| Elimination | |
| CYP1A2 | Vmax: 7.928, Km: 20.777 c |
| CYP2D6 | Vmax: 2.319, Km: 14.602 c |
| Transporter | |
| p-gp (ABCB1) | Clint: 18 d |
| Starting Dose | 6 h | 24 h | 48 h | |
|---|---|---|---|---|
| Reduced dose 1 | 465 mg | 310 mg | 310 mg | 310 mg |
| Reduced dose 2 | 465 mg | 155 mg | 310 mg | 310 mg |
| Reduced dose 3 | 465 mg | 310 mg | 155 mg | 310 mg |
| Reduced dose 4 | 465 mg | 310 mg | 310 mg | 155 mg |
| Reduced dose 5 | 465 mg | 155 mg | 310 mg | 155 mg |
| Reduced dose 6 | 465 mg | 310 mg | 155 mg | 155 mg |
| MO-First Dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.23 | 0.22 | 0.23 | 0.23 | 0.23 |
| GFR | 0.04 | 0.04 | 0.04 | 0.05 | 0.04 |
| Height | 0.19 | 0.20 | 0.20 | 0.20 | 0.19 |
| Serum creatine | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| MO-Second dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.30 | 0.29 | 0.30 | 0.30 | 0.30 |
| GFR | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 |
| Height | 0.29 | 0.29 | 0.29 | 0.29 | 0.29 |
| Serum creatine | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 |
| MO-Third dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.25 | 0.24 | 0.25 | 0.26 | 0.25 |
| GFR | 0.09 | 0.09 | 0.09 | 0.09 | 0.08 |
| Height | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 |
| Serum creatine | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| MO-Fourth dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.21 | 0.20 | 0.21 | 0.22 | 0.21 |
| GFR | 0.11 | 0.11 | 0.10 | 0.11 | 0.09 |
| Height | 0.19 | 0.19 | 0.19 | 0.19 | 0.19 |
| Serum creatine | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| Pediatric-First Dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.68 | 0.52 | 0.76 | 0.71 | 0.90 |
| Height | 0.86 | 0.75 | 0.82 | 0.85 | 0.87 |
| Age | 0.72 | 0.59 | 0.83 | 0.79 | 0.85 |
| GFR | 0.66 | 0.50 | 0.71 | 0.73 | 0.79 |
| Pediatric-Second dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.71 | 0.53 | 0.78 | 0.73 | 0.93 |
| Height | 0.89 | 0.78 | 0.84 | 0.88 | 0.89 |
| Age | 0.76 | 0.62 | 0.87 | 0.82 | 0.87 |
| GFR | 0.70 | 0.53 | 0.74 | 0.76 | 0.83 |
| Pediatric-Third dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.73 | 0.55 | 0.78 | 0.74 | 0.90 |
| Height | 0.85 | 0.77 | 0.82 | 0.84 | 0.83 |
| Age | 0.77 | 0.63 | 0.84 | 0.80 | 0.80 |
| GFR | 0.71 | 0.54 | 0.74 | 0.76 | 0.79 |
| Pediatric-Fourth dose | |||||
| Exponential | Linear | Logarithmic | Polynomial | Power | |
| Weight | 0.71 | 0.54 | 0.76 | 0.72 | 0.86 |
| Height | 0.81 | 0.74 | 0.79 | 0.80 | 0.79 |
| Age | 0.73 | 0.61 | 0.81 | 0.76 | 0.77 |
| GFR | 0.69 | 0.53 | 0.72 | 0.74 | 0.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, J.; Ji, B.; Cai, L.; Liu, S.; Sun, Y.; Wang, J. Physiologically-Based Pharmacokinetics Modeling for Hydroxychloroquine as a Treatment for Malaria and Optimized Dosing Regimens for Different Populations. J. Pers. Med. 2022, 12, 796. https://doi.org/10.3390/jpm12050796
Zhai J, Ji B, Cai L, Liu S, Sun Y, Wang J. Physiologically-Based Pharmacokinetics Modeling for Hydroxychloroquine as a Treatment for Malaria and Optimized Dosing Regimens for Different Populations. Journal of Personalized Medicine. 2022; 12(5):796. https://doi.org/10.3390/jpm12050796
Chicago/Turabian StyleZhai, Jingchen, Beihong Ji, Lianjin Cai, Shuhan Liu, Yuchen Sun, and Junmei Wang. 2022. "Physiologically-Based Pharmacokinetics Modeling for Hydroxychloroquine as a Treatment for Malaria and Optimized Dosing Regimens for Different Populations" Journal of Personalized Medicine 12, no. 5: 796. https://doi.org/10.3390/jpm12050796
APA StyleZhai, J., Ji, B., Cai, L., Liu, S., Sun, Y., & Wang, J. (2022). Physiologically-Based Pharmacokinetics Modeling for Hydroxychloroquine as a Treatment for Malaria and Optimized Dosing Regimens for Different Populations. Journal of Personalized Medicine, 12(5), 796. https://doi.org/10.3390/jpm12050796