
FSHD Therapeutic Strategies: What Will It Take to Get to Clinic?

{kind=link}
{kind=link}
{kind=link}
Abstract
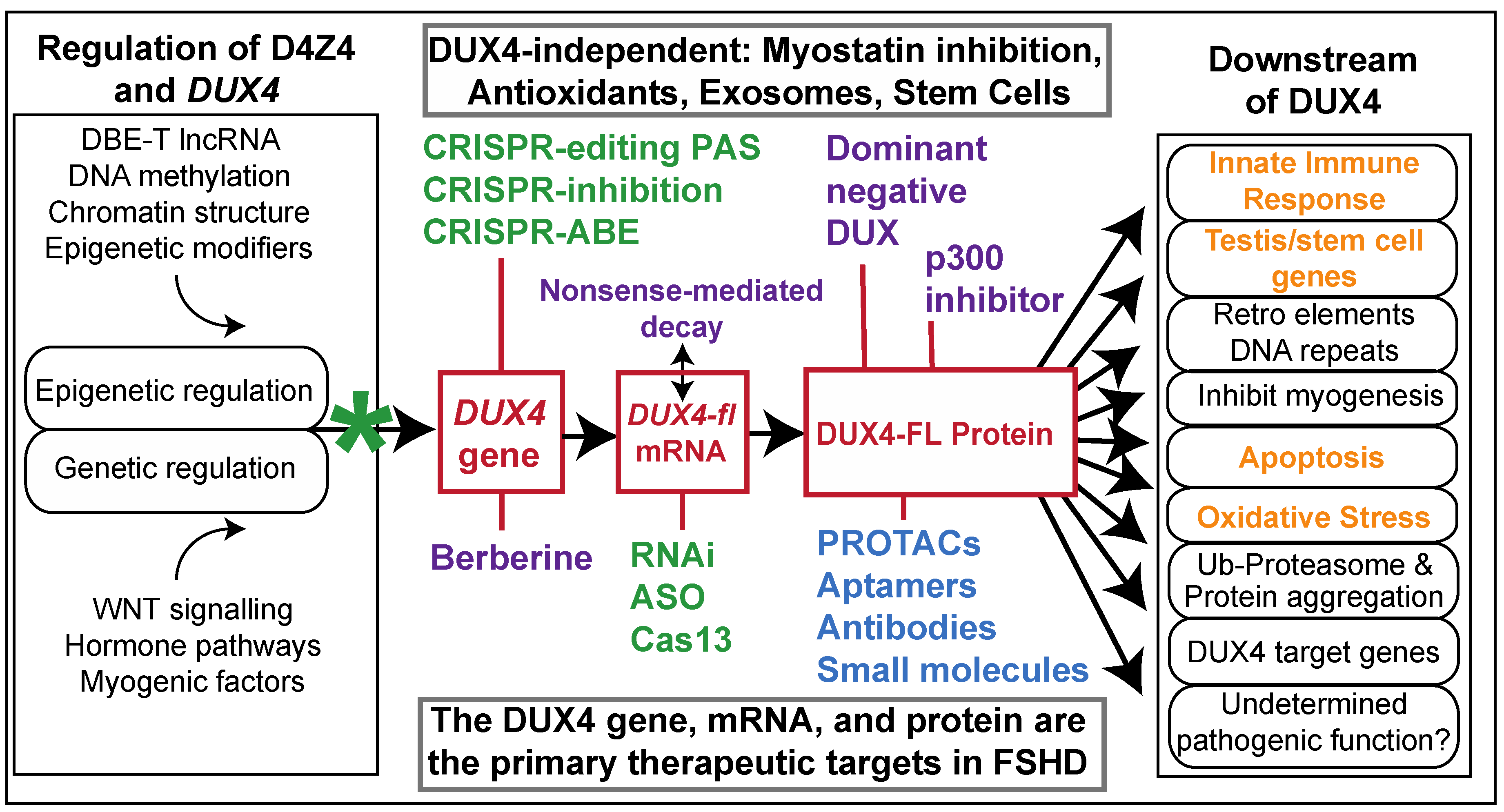
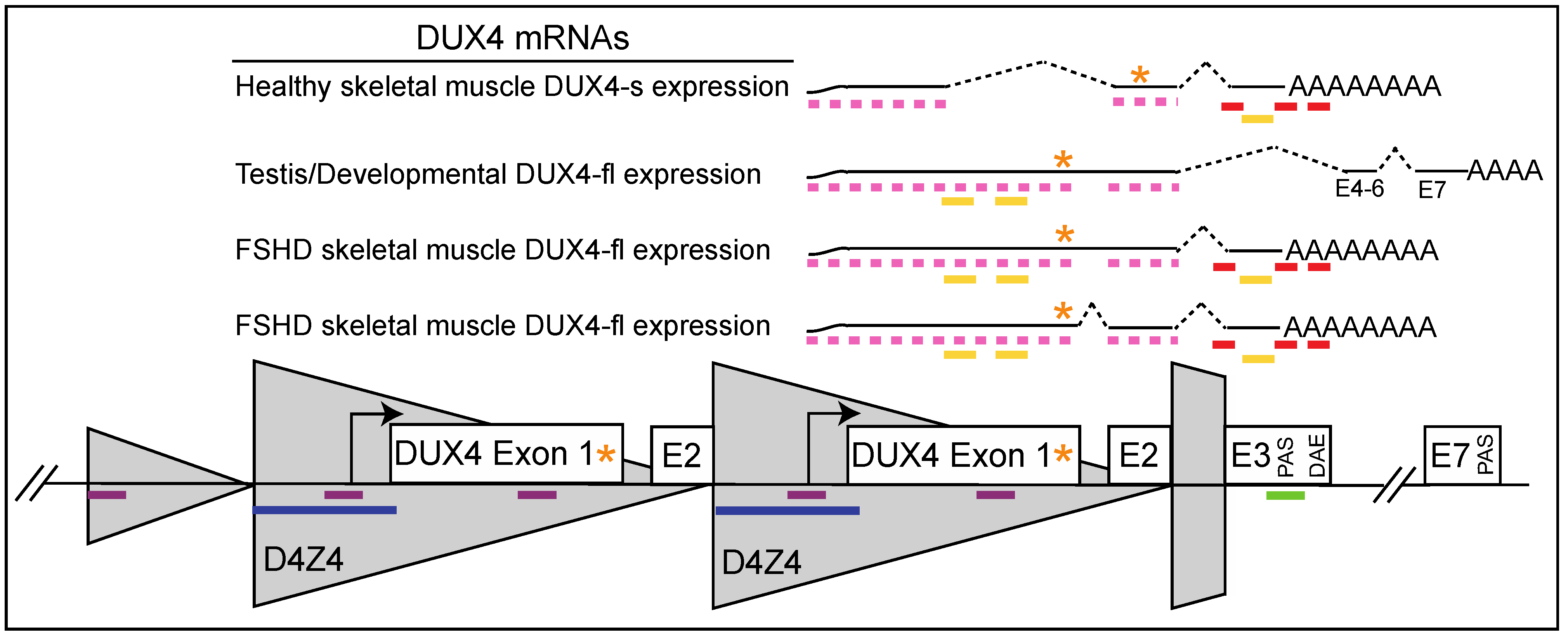
:1. Introduction
2. Small-Molecule Drugs
2.1. Progress in the Field
2.2. Advantages
2.3. Disadvantages
2.4. Path to Clinic
3. Oligonucleotide Therapeutics
3.1. Progress in the Field
3.2. Advantages
- -
- ability to directly repress expression of DUX4, a difficult-to-drug transcription factor
- -
- relatively straightforward to design, produce, and screen
- -
- sequence-based targeting allows for precision/personalized medicine
- -
- treatment can be stopped if adverse events occur
3.3. Disadvantages
3.4. Path to Clinic
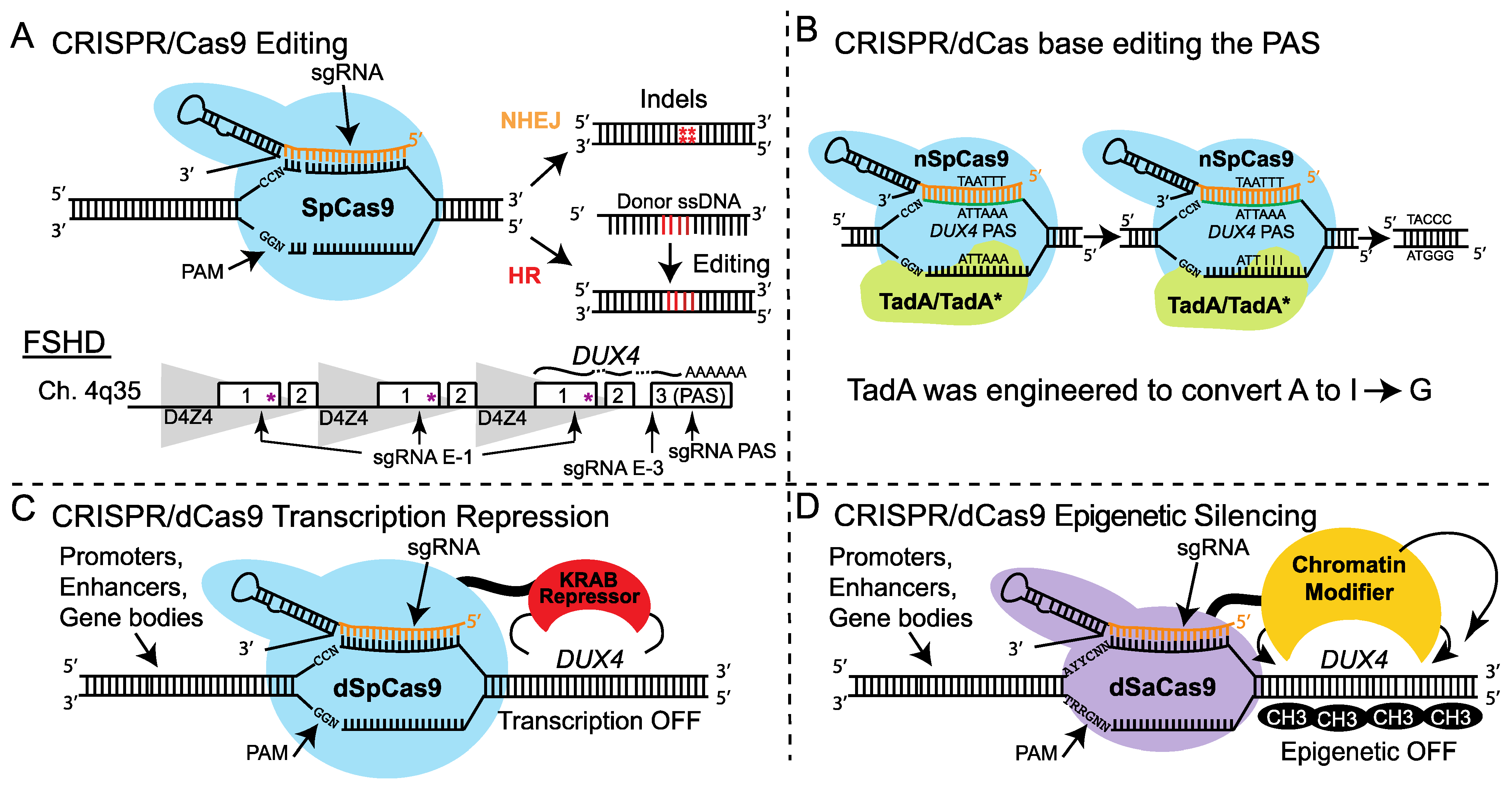
4. Gene Modification
4.1. Progress in the Field
4.2. Advantages
- -
- Both CRISPRe and CRISPRi have the potential to achieve long-term or permanent correction of all forms of FSHD with a single treatment.
- -
- In principle, CRISPRi is ideally suited to a dominant, gain-of-function disease such as FSHD. Since it does not involve cutting the genome, CRISPRi may also be safer than CRISPRe.
- -
- In the long run, CRISPR therapies (and other gene therapy approaches) may be the most economical means of treatment, since they are delivered in a single dose that is meant to function over a lifetime.
4.3. Disadvantages
4.4. Path to Clinic
- -
- For CRISPRe, rigorous tests of specificity are required. Rapid advances in specific, programmable base-editing (e.g., prime-editing) [89] should greatly improve the safety of CRISPRe for FSHD. However, current base editors are much too large to be accommodated in a single-vector system for rAAV-mediated delivery [89]. These need to be better characterized and minimized to be therapeutically relevant.
- -
- Stable repression mediated by CRISPRi remains to be demonstrated and is absolutely necessary to achieve, since current rAAV vectors for gene therapy can only be administered once. In brief, either the CRISPRi treatment must elicit a stable epigenetic change or the rAAV episome must persist indefinitely in treated fibers (discussed in more detail below).
- -
- Both types of CRISPR approaches must be tested more rigorously in appropriate in vivo models, including a large animal model (see below). For CRISPRi, a human xenograft model [90,91] is ideal for assessing the persistence of epigenetic changes at the disease locus, as xenografted mice contain a full D4Z4 array from an FSHD patient.
5. Needs for Therapeutics Delivered by Gene Therapy (CRISPR Strategies, RNAi)
5.1. Delivery to Body-Wide Skeletal Muscles
5.2. Overcoming rAAV-Related Limitations
6. Needs for All DUX4-Targeted Therapeutics
6.1. A Large Animal Model of FSHD for Preclinical Testing of Therapeutics
6.2. DUX4-Responsive Circulating Biomarkers of Disease Progression
7. DUX4-Independent Approaches
8. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Padberg, G.W. Facioscapulohumeral Disease. Ph.D. Thesis, Leiden University, Leiden, The Netherlands, 1982. [Google Scholar]
- Deenen, J.C.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.; van Engelen, B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66. [Google Scholar] [CrossRef]
- Orphanet. Prevalence and Incidence of Rare Diseases: Bibliographic Data. Orphanet Report Series: Rare Diseases Collection. 2021. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf (accessed on 23 May 2022).
- Tawil, R.; Van Der Maarel, S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Deenen, J.C.W.; Voermans, N.C.; van Engelen, B.G.M.; Kievit, W.; Groothuis, J.T. The socioeconomic burden of facioscapulohumeral muscular dystrophy. J. Neurol. 2021, 268, 4778–4788. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Hendrickson, P.G.; Dorais, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- Whiddon, J.L.; Langford, A.T.; Wong, C.J.; Zhong, J.W.; Tapscott, S.J. Conservation and innovation in the DUX4-family gene network. Nat. Genet. 2017, 49, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Mbakam, C.H.; Lamothe, G.; Tremblay, J.P. Therapeutic Strategies for Dystrophin Replacement in Duchenne Muscular Dystrophy. Front. Med. 2022, 9, 859930. [Google Scholar] [CrossRef]
- Pozsgai, E.; Griffin, D.; Potter, R.; Sahenk, Z.; Lehman, K.; Rodino-Klapac, L.R.; Mendell, J.R. Unmet needs and evolving treatment for limb girdle muscular dystrophies. Neurodegener. Dis. Manag. 2021, 11, 411–429. [Google Scholar] [CrossRef]
- Soltanzadeh, P. Myotonic Dystrophies: A Genetic Overview. Genes 2022, 13, 367. [Google Scholar] [CrossRef]
- Butterfield, R.J. Congenital Muscular Dystrophy and Congenital Myopathy. Continuum 2019, 25, 1640–1661. [Google Scholar] [CrossRef]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448. [Google Scholar] [CrossRef]
- Lyle, R.; Wright, T.J.; Clark, L.N.; Hewitt, J.E. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics 1995, 28, 389–397. [Google Scholar] [CrossRef]
- Gabriels, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.; de Kievit, P.; van Geel, M.; van der Wielen, M.J.; Bakker, E.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann. Neurol. 2001, 50, 816–819. [Google Scholar] [CrossRef]
- Lemmers, R.J.; de Kievit, P.; Sandkuijl, L.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002, 32, 235–236. [Google Scholar] [CrossRef]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.I.; Chen, J.C.; Rahimov, F.; Homma, S.; Arashiro, P.; Beermann, M.L.; King, O.D.; Miller, J.B.; Kunkel, L.M.; Emerson, C.P., Jr.; et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: Evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012, 21, 4419–4430. [Google Scholar] [CrossRef] [Green Version]
- Himeda, C.L.; Debarnot, C.; Homma, S.; Beermann, M.L.; Miller, J.B.; Jones, P.L.; Jones, T.I. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy associated DUX4 gene. Mol. Cell. Biol. 2014, 34, 1942–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Clapp, J.; Mitchell, L.M.; Bolland, D.J.; Fantes, J.; Corcoran, A.E.; Scotting, P.J.; Armour, J.A.; Hewitt, J.E. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007, 81, 264–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidenroth, A.; Clapp, J.; Mitchell, L.M.; Coneyworth, D.; Dearden, F.L.; Iannuzzi, L.; Hewitt, J.E. Evolution of DUX gene macrosatellites in placental mammals. Chromosoma 2012, 121, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Leidenroth, A.; Hewitt, J.E. A family history of DUX4: Phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol. Biol. 2010, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Lek, A.; Rahimov, F.; Jones, P.L.; Kunkel, L.M. Emerging preclinical animal models for FSHD. Trends Mol. Med. 2015, 21, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.C.; Lochmuller, H.; Reilich, P.; Klopstock, T.; Huber, R.; Hartard, M.; Hennig, M.; Pongratz, D.; Muller-Felber, W. Creatine monohydrate in muscular dystrophies: A double-blind, placebo-controlled clinical study. Neurology 2000, 54, 1848–1850. [Google Scholar] [CrossRef]
- Kissel, J.T.; McDermott, M.P.; Mendell, J.R.; King, W.M.; Pandya, S.; Griggs, R.C.; Tawil, R.; Group, F.-D. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology 2001, 57, 1434–1440. [Google Scholar] [CrossRef]
- van der Kooi, E.L.; de Greef, J.C.; Wohlgemuth, M.; Frants, R.R.; van Asseldonk, R.J.; Blom, H.J.; van Engelen, B.G.; van der Maarel, S.M.; Padberg, G.W. No effect of folic acid and methionine supplementation on D4Z4 methylation in patients with facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2006, 16, 766–769. [Google Scholar] [CrossRef] [Green Version]
- van der Kooi, E.L.; Kalkman, J.S.; Lindeman, E.; Hendriks, J.C.; van Engelen, B.G.; Bleijenberg, G.; Padberg, G.W. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J. Neurol. 2007, 254, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Glasser, C.E.; Gartner, M.R.; Wilson, D.; Miller, B.; Sherman, M.L.; Attie, K.M. Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve 2018, 57, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.C.; Bocker, K.; Hugon, G.; Pincemail, J.; et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Campbell, C.; Desai, U.; Karam, C.; Diaz-Manera, J.; Guptill, J.T.; Korngut, L.; Genge, A.; Tawil, R.N.; Elman, L.; et al. Randomized Phase 2 Study of ACE-083, a Muscle-Promoting Agent, in Facioscapulohumeral Muscular Dystrophy. Muscle Nerve, 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Mellion, M.L.; Ronco, L.; Berends, C.L.; Pagan, L.; Brooks, S.; van Esdonk, M.J.; van Brummelen, E.M.J.; Odueyungbo, A.; Thompson, L.A.; Hage, M.; et al. Phase 1 clinical trial of losmapimod in facioscapulohumeral dystrophy: Safety, tolerability, pharmacokinetics, and target engagement. Br. J. Clin. Pharmacol. 2021, 87, 4658–4669. [Google Scholar] [CrossRef]
- Pascual-Gilabert, M.; Lopez-Castel, A.; Artero, R. Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discov. Today 2021, 26, 1765–1772. [Google Scholar] [CrossRef]
- Le Gall, L.; Sidlauskaite, E.; Mariot, V.; Dumonceaux, J. Therapeutic Strategies Targeting DUX4 in FSHD. J. Clin. Med. 2020, 9, 2886. [Google Scholar] [CrossRef]
- Wang, L.H.; Tawil, R. Current Therapeutic Approaches in FSHD. J. Neuromuscul. Dis. 2021, 8, 441–451. [Google Scholar] [CrossRef]
- Cohen, J.; DeSimone, A.; Lek, M.; Lek, A. Therapeutic Approaches in Facioscapulohumeral Muscular Dystrophy. Trends Mol. Med. 2021, 27, 123–137. [Google Scholar] [CrossRef]
- Campbell, A.E.; Oliva, J.; Yates, M.P.; Zhong, J.W.; Shadle, S.C.; Snider, L.; Singh, N.; Tai, S.; Hiramuki, Y.; Tawil, R.; et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle 2017, 7, 16. [Google Scholar] [CrossRef]
- Cruz, J.M., Jr.; Hupper, N.; Wilson, L.S.; Concannon, J.B.; Wang, Y.; Oberhauser, B.; Patora-Komisarska, K.; Zhang, Y.; Glass, D.J.; Trendelenburg, A.U.; et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J. Biol. Chem. 2018, 293, 11837–11849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, L.A.; Valentine, E.; Accorsi, A.; Maglio, J.; Shen, N.; Robertson, A.; Kazmirski, S.; Rahl, P.; Tawil, R.; Cadavid, D.; et al. P38 alpha Regulates Expression of DUX4 in a Model of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2020, 374, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Block, G.J.; Narayanan, D.; Amell, A.M.; Petek, L.M.; Davidson, K.C.; Bird, T.D.; Tawil, R.; Moon, R.T.; Miller, D.G. Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 2013, 22, 4661–4672. [Google Scholar] [CrossRef] [Green Version]
- Himeda, C.L.; Jones, T.I.; Virbasius, C.M.; Zhu, L.J.; Green, M.R.; Jones, P.L. Identification of Epigenetic Regulators of DUX4-fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2018, 26, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.E.; Shadle, S.C.; Jagannathan, S.; Lim, J.W.; Resnick, R.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. eLife 2018, 7, e31023. [Google Scholar] [CrossRef]
- Sharma, V.; Pandey, S.N.; Khawaja, H.; Brown, K.J.; Hathout, Y.; Chen, Y.W. PARP1 Differentially Interacts with Promoter region of DUX4 Gene in FSHD Myoblasts. J. Genet. Syndr. Gene Ther. 2016, 7, 303. [Google Scholar] [CrossRef]
- Balog, J.; Thijssen, P.E.; Shadle, S.; Straasheijm, K.R.; van der Vliet, P.J.; Krom, Y.D.; van den Boogaard, M.L.; de Jong, A.; Lemmers, R.J.L.F.; Tawil, R.; et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics 2015, 10, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Lek, A.; Zhang, Y.; Woodman, K.G.; Huang, S.; DeSimone, A.M.; Cohen, J.; Ho, V.; Conner, J.; Mead, L.; Kodani, A.; et al. Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 2020, 12, eaay0271. [Google Scholar] [CrossRef]
- Choi, S.H.; Bosnakovski, D.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. Transcriptional Inhibitors Identified in a 160,000-Compound Small-Molecule DUX4 Viability Screen. J. Biomol. Screen. 2016, 21, 680–688. [Google Scholar] [CrossRef] [Green Version]
- Bosnakovski, D.; Choi, S.H.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet. Muscle 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makurvet, F.D. Biologics vs. small molecules: Drug costs and patient access. Med. Drug Discov. 2021, 9, 100075. [Google Scholar] [CrossRef]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef]
- Wissing, E.R.; Boyer, J.G.; Kwong, J.Q.; Sargent, M.A.; Karch, J.; McNally, E.M.; Otsu, K.; Molkentin, J.D. P38alpha MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 2014, 23, 5452–5463. [Google Scholar] [CrossRef] [Green Version]
- Segales, J.; Perdiguero, E.; Munoz-Canoves, P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91. [Google Scholar] [CrossRef] [Green Version]
- Rask-Andersen, M.; Zhang, J.; Fabbro, D.; Schioth, H.B. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol. Sci. 2014, 35, 604–620. [Google Scholar] [CrossRef]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppee, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS ONE 2011, 6, e26820. [Google Scholar] [CrossRef]
- Ansseau, E.; Vanderplanck, C.; Wauters, A.; Harper, S.Q.; Coppee, F.; Belayew, A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes 2017, 8, 93. [Google Scholar] [CrossRef]
- Lim, J.W.; Snider, L.; Yao, Z.; Tawil, R.; Van Der Maarel, S.M.; Rigo, F.; Bennett, C.F.; Filippova, G.N.; Tapscott, S.J. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum. Mol. Genet. 2015, 24, 4817–4828. [Google Scholar] [CrossRef] [Green Version]
- Marsollier, A.C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum. Mol. Genet. 2016, 25, 1468–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.R.Q.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Zhang, A.; Khawaja, H.; Sen Chandra, S.; Jones, T.; Jones, P.; Chen, Y.W.; et al. Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 16509–16515. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Bittel, A.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Huang, Y.; Dzierlega, K.; Zhang, A.; Chen, Y.W.; Yokota, T. DUX4 Transcript Knockdown with Antisense 2′-O-Methoxyethyl Gapmers for the Treatment of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2021, 29, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Malerba, A.; Pineda, M.A.; Dickson, G.; Popplewell, L.J. Improving molecular and histopathology in diaphragm muscle of the double transgenic ACTA1-MCM/FLExDUX4 mouse model of FSHD with systemic antisense therapy. Hum. Gene Ther. 2022; ahead of print. [Google Scholar] [CrossRef]
- Lu-Nguyen, N.; Malerba, A.; Herath, S.; Dickson, G.; Popplewell, L. Systemic antisense therapeutics inhibiting DUX4 expression ameliorates FSHD-like pathology in an FSHD mouse model. Hum. Mol. Genet. 2021, 30, 1398–1412. [Google Scholar] [CrossRef]
- Wallace, L.M.; Liu, J.; Domire, J.S.; Garwick-Coppens, S.E.; Guckes, S.M.; Mendell, J.R.; Flanigan, K.M.; Harper, S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: Implications for a targeted FSHD therapy. Mol. Ther. 2012, 20, 1417–1423. [Google Scholar] [CrossRef] [Green Version]
- Wallace, L.M.; Saad, N.Y.; Pyne, N.K.; Fowler, A.M.; Eidahl, J.O.; Domire, J.S.; Griffin, D.A.; Herman, A.C.; Sahenk, Z.; Rodino-Klapac, L.R.; et al. Pre-clinical Safety and Off-Target Studies to Support Translation of AAV-Mediated RNAi Therapy for FSHD. Mol. Ther. Methods Clin. Dev. 2018, 8, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Rashnonejad, A.; Amini-Chermahini, G.; Taylor, N.K.; Wein, N.; Harper, S.Q. Designed U7 snRNAs inhibit DUX4 expression and improve FSHD-associated outcomes in DUX4 overexpressing cells and FSHD patient myotubes. Mol. Ther. Nucleic Acids 2021, 23, 476–486. [Google Scholar] [CrossRef]
- Klingler, C.; Ashley, J.; Shi, K.; Stiefvater, A.; Kyba, M.; Sinnreich, M.; Aihara, H.; Kinter, J. DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. FASEB J. 2020, 34, 4573–4590. [Google Scholar] [CrossRef] [Green Version]
- Mariot, V.; Joubert, R.; Marsollier, A.C.; Hourdé, C.; Voit, T.; Dumonceaux, J. A Deoxyribonucleic Acid Decoy Trapping DUX4 for the Treatment of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. Nucleic Acids 2020, 22, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Nichols, J.G.; De Hoyos, C.L.; Crooke, S.T. Some ASOs that bind in the coding region of mRNAs and induce RNase H1 cleavage can cause increases in the pre-mRNAs that may blunt total activity. Nucleic Acids Res. 2020, 48, 9840–9858. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Joubert, R.; Mariot, V.; Charpentier, M.; Concordet, J.P.; Dumonceaux, J. Gene Editing Targeting the DUX4 Polyadenylation Signal: A Therapy for FSHD? J. Pers. Med. 2020, 11, 7. [Google Scholar] [CrossRef]
- Das, S.; Chadwick, B.P. CRISPR mediated targeting of DUX4 distal regulatory element represses DUX4 target genes dysregulated in Facioscapulohumeral muscular dystrophy. Sci. Rep. 2021, 11, 12598. [Google Scholar] [CrossRef]
- Sikrova, D.; Cadar, V.A.; Ariyurek, Y.; Laros, J.F.J.; Balog, J.; van der Maarel, S.M. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids 2021, 25, 342–354. [Google Scholar] [CrossRef]
- Goossens, R.; van den Boogaard, M.L.; Lemmers, R.; Balog, J.; van der Vliet, P.J.; Willemsen, I.M.; Schouten, J.; Maggio, I.; van der Stoep, N.; Hoeben, R.C.; et al. Intronic SMCHD1 variants in FSHD: Testing the potential for CRISPR-Cas9 genome editing. J. Med. Genet. 2019, 56, 828–837. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol. Ther. 2016, 24, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Scalpel or Straitjacket: CRISPR/Cas9 Approaches for Muscular Dystrophies. Trends Pharamacol. Sci. 2016, 37, 249–251. [Google Scholar] [CrossRef] [Green Version]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol. Ther. Methods Clin. Dev. 2021, 20, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Merkel, O.M. Immunogenicity of Cas9 Protein. J. Pharm. Sci. 2020, 109, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019, 10, 1842. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Venneri, M.A.; Zingale, A.; Sergi, L.; Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006, 12, 585–591. [Google Scholar] [CrossRef]
- Muhuri, M.; Zhan, W.; Maeda, Y.; Li, J.; Lotun, A.; Chen, J.; Sylvia, K.; Dasgupta, I.; Arjomandnejad, M.; Nixon, T.; et al. Novel Combinatorial MicroRNA-Binding Sites in AAV Vectors Synergistically Diminish Antigen Presentation and Transgene Immunity for Efficient and Stable Transduction. Front. Immunol. 2021, 12, 674242. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Sakellariou, P.; O’Neill, A.; Mueller, A.L.; Stadler, G.; Wright, W.E.; Roche, J.S.; Bloch, R.J. Neuromuscular electrical stimulation promotes development in mice of mature human muscle from immortalized human myoblasts. Skelet. Muscle 2016, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.L.; O’Neill, A.; Jones, T.I.; Llach, A.; Rojas, L.A.; Sakellariou, P.; Stadler, G.; Wright, W.E.; Eyerman, D.; Jones, P.L.; et al. Muscle xenografts reproduce key molecular features of facioscapulohumeral muscular dystrophy. Exp. Neurol. 2019, 320, 113011. [Google Scholar] [CrossRef]
- Hareendran, S.; Balakrishnan, B.; Sen, D.; Kumar, S.; Srivastava, A.; Jayandharan, G.R. Adeno-associated virus (AAV) vectors in gene therapy: Immune challenges and strategies to circumvent them. Rev. Med. Virol. 2013, 23, 399–413. [Google Scholar] [CrossRef]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938.e22. [Google Scholar] [CrossRef] [PubMed]
- Faust, S.M.; Bell, P.; Cutler, B.J.; Ashley, S.N.; Zhu, Y.; Rabinowitz, J.E.; Wilson, J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Investig. 2013, 123, 2994–3001. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Verdera, H.C.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J., Jr.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13, eabd3438. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Cherel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Chu, W.S.; Ng, J. Immunomodulation in Administration of rAAV: Preclinical and Clinical Adjuvant Pharmacotherapies. Front. Immunol. 2021, 12, 658038. [Google Scholar] [CrossRef]
- Bertin, B.; Veron, P.; Leborgne, C.; Deschamps, J.Y.; Moullec, S.; Fromes, Y.; Collaud, F.; Boutin, S.; Latournerie, V.; van Wittenberghe, L.; et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci. Rep. 2020, 10, 864. [Google Scholar] [CrossRef] [Green Version]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Maheshri, N.; Kaspar, B.; Schaffer, D.V. PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 2005, 92, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Huttner, N.A.; Girod, A.; Perabo, L.; Edbauer, D.; Kleinschmidt, J.A.; Buning, H.; Hallek, M. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther. 2003, 10, 2139–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.; Ragni, M.V.; Manno, C.S.; Sommer, J.; Jiang, H.; et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef]
- Mueller, C.; Chulay, J.D.; Trapnell, B.C.; Humphries, M.; Carey, B.; Sandhaus, R.A.; McElvaney, N.G.; Messina, L.; Tang, Q.; Rouhani, F.N.; et al. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J. Clin. Investig. 2013, 123, 5310–5318. [Google Scholar] [CrossRef]
- Biswas, M.; Kumar, S.R.P.; Terhorst, C.; Herzog, R.W. Gene Therapy With Regulatory T Cells: A Beneficial Alliance. Front. Immunol. 2018, 9, 554. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.F. AAV empty capsids: For better or for worse? Mol. Ther. 2014, 22, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.; Gambhir, Y.; Bennett, J.; Stedman, H.H. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Mol. Ther. 2020, 28, 1753–1755. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Xie, J.; Mao, Q.; Tai, P.W.L.; He, R.; Ai, J.; Su, Q.; Zhu, Y.; Ma, H.; Li, J.; Gong, S.; et al. Short DNA Hairpins Compromise Recombinant Adeno-Associated Virus Genome Homogeneity. Mol. Ther. 2017, 25, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.T.; Heiner, C.; Weber, K.; Weiand, M.; Wilmot, D.; Xie, J.; Wang, D.; Brown, A.; Manokaran, S.; Su, Q.; et al. AAV-Genome Population Sequencing of Vectors Packaging CRISPR Components Reveals Design-Influenced Heterogeneity. Mol. Ther. Methods Clin. Dev. 2020, 18, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Eddington, C.; Jose, A.; Hsi, J.; Chipman, P.; Henley, T.; Choudhry, M.; McKenna, R.; Agbandje-McKenna, M. Improved Genome Packaging Efficiency of Adeno-associated Virus Vectors Using Rep Hybrids. J. Virol. 2021, 95, e0077321. [Google Scholar] [CrossRef]
- Echigoya, Y.; Trieu, N.; Duddy, W.; Moulton, H.M.; Yin, H.; Partridge, T.A.; Hoffman, E.P.; Kornegay, J.N.; Rohret, F.A.; Rogers, C.S.; et al. A Dystrophin Exon-52 Deleted Miniature Pig Model of Duchenne Muscular Dystrophy and Evaluation of Exon Skipping. Int. J. Mol. Sci. 2021, 22, 13065. [Google Scholar] [CrossRef]
- Valles, A.; Evers, M.M.; Stam, A.; Sogorb-Gonzalez, M.; Brouwers, C.; Vendrell-Tornero, C.; Acar-Broekmans, S.; Paerels, L.; Klima, J.; Bohuslavova, B.; et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci. Transl. Med. 2021, 13, eabb8920. [Google Scholar] [CrossRef] [PubMed]
- Schachtschneider, K.M.; Schook, L.B.; Meudt, J.J.; Shanmuganayagam, D.; Zoller, J.A.; Haghani, A.; Li, C.Z.; Zhang, J.; Yang, A.; Raj, K.; et al. Epigenetic clock and DNA methylation analysis of porcine models of aging and obesity. Geroscience 2021, 43, 2467–2483. [Google Scholar] [CrossRef]
- Swindle, M.M. The development of swine models in drug discovery and development. Future Med. Chem. 2012, 4, 1771–1772. [Google Scholar] [CrossRef] [PubMed]
- Swindle, M.M.; Makin, A.; Herron, A.J.; Clubb, F.J., Jr.; Frazier, K.S. Swine as models in biomedical research and toxicology testing. Vet. Pathol. 2012, 49, 344–356. [Google Scholar] [CrossRef]
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the porcine species in biomedical research. Front. Genet. 2015, 6, 293. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.I.; Chew, G.L.; Barraza-Flores, P.; Schreier, S.; Ramirez, M.; Wuebbles, R.D.; Burkin, D.J.; Bradley, R.K.; Jones, P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle 2020, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.; Donlin-Smith, C.M.; Tapscott, S.J.; van der Maarel, S.; Tawil, R. Multiplex Screen of Serum Biomarkers in Facioscapulohumeral Muscular Dystrophy. J. Neuromuscul. Dis. 2014, 1, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portilho, D.M.; Alves, M.R.; Kratassiouk, G.; Roche, S.; Magdinier, F.; de Santana, E.C.; Polesskaya, A.; Harel-Bellan, A.; Mouly, V.; Savino, W.; et al. miRNA Expression in Control and FSHD Fetal Human Muscle Biopsies. PLoS ONE 2015, 10, e0116853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petek, L.M.; Rickard, A.M.; Budech, C.; Poliachik, S.L.; Shaw, D.; Ferguson, M.R.; Tawil, R.; Friedman, S.D.; Miller, D.G. A cross sectional study of two independent cohorts identifies serum biomarkers for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2016, 26, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heier, C.R.; Zhang, A.; Nguyen, N.Y.; Tully, C.B.; Panigrahi, A.; Gordish-Dressman, H.; Pandey, S.N.; Guglieri, M.; Ryan, M.M.; Clemens, P.R.; et al. Multi-Omics Identifies Circulating miRNA and Protein Biomarkers for Facioscapulohumeral Dystrophy. J. Pers. Med. 2020, 10, 236. [Google Scholar] [CrossRef]
- Nunes, A.M.; Ramirez, M.; Jones, T.I.; Jones, P.L. Identification of candidate miRNA biomarkers for facioscapulohumeral muscular dystrophy using DUX4-based mouse models. Dis. Models Mech. 2021, 14, dmm049016. [Google Scholar] [CrossRef]
- Gros, M.; Nunes, A.M.; Daoudlarian, D.; Pini, J.; Martinuzzi, E.; Barbosa, S.; Ramirez, M.; Puma, A.; Villa, L.; Cavalli, M.; et al. Identification of Serum Interleukin 6 Levels as a Disease Severity Biomarker in Facioscapulohumeral Muscular Dystrophy. J. Neuromuscul. Dis. 2022, 9, 83–93. [Google Scholar] [CrossRef]
- Ghasemi, M.; Emerson, C.P., Jr.; Hayward, L.J. Outcome Measures in Facioscapulohumeral Muscular Dystrophy Clinical Trials. Cells 2022, 11, 687. [Google Scholar] [CrossRef]
- Butz, M.; Koch, M.C.; Muller-Felber, W.; Lemmers, R.J.; van der Maarel, S.M.; Schreiber, H. Facioscapulohumeral muscular dystrophy. Phenotype-genotype correlation in patients with borderline D4Z4 repeat numbers. J. Neurol. 2003, 250, 932–937. [Google Scholar] [CrossRef]
- Salort-Campana, E.; Nguyen, K.; Bernard, R.; Jouve, E.; Sole, G.; Nadaj-Pakleza, A.; Niederhauser, J.; Charles, E.; Ollagnon, E.; Bouhour, F.; et al. Low penetrance in facioscapulohumeral muscular dystrophy type 1 with large pathological D4Z4 alleles: A cross-sectional multicenter study. Orphanet J. Rare Dis. 2015, 10, 2. [Google Scholar] [CrossRef]
- He, J.J.; Lin, X.D.; Lin, F.; Xu, G.R.; Xu, L.Q.; Hu, W.; Wang, D.N.; Lin, H.X.; Lin, M.T.; Wang, N.; et al. Clinical and genetic features of patients with facial-sparing facioscapulohumeral muscular dystrophy. Eur. J. Neurol. 2018, 25, 356–364. [Google Scholar] [CrossRef]
- Ruggiero, L.; Mele, F.; Manganelli, F.; Bruzzese, D.; Ricci, G.; Vercelli, L.; Govi, M.; Vallarola, A.; Tripodi, S.; Villa, L.; et al. Phenotypic Variability Among Patients With D4Z4 Reduced Allele Facioscapulohumeral Muscular Dystrophy. JAMA Netw. Open 2020, 3, e204040. [Google Scholar] [CrossRef] [PubMed]
- Heher, P.; Ganassi, M.; Weidinger, A.; Engquist, E.N.; Pruller, J.; Nguyen, T.H.; Tassin, A.; Decleves, A.E.; Mamchaoui, K.; Banerji, C.R.S.; et al. Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: Metabolic stress as potential therapeutic target. Redox Biol. 2022, 51, 102251. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; MacDonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, A.I.; Nagy, P.L.; Manta, K.; Rouse, N.; Manta, A.; Ng, S.Y.; Nagy, M.F.; Smith, P.; Lu, J.Q.; Nederveen, J.P.; et al. Aerobic exercise elicits clinical adaptations in myotonic dystrophy type 1 patients independent of pathophysiological changes. J. Clin. Investig. 2022, 132, e156125. [Google Scholar] [CrossRef]
- Kley, R.A.; Tarnopolsky, M.A.; Vorgerd, M. Creatine treatment in muscle disorders: A meta-analysis of randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2008, 79, 366–367. [Google Scholar] [CrossRef]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R. The elusive promise of myostatin inhibition for muscular dystrophy. Curr. Opin. Neurol. 2020, 33, 621–628. [Google Scholar] [CrossRef]
- Willmann, R.; Lee, J.; Turner, C.; Nagaraju, K.; Aartsma-Rus, A.; Wells, D.J.; Wagner, K.R.; Csimma, C.; Straub, V.; Grounds, M.D.; et al. Improving translatability of preclinical studies for neuromuscular disorders: Lessons from the TREAT-NMD Advisory Committee for Therapeutics (TACT). Dis. Models Mech. 2020, 13, dmm042903. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.I.; King, O.D.; Himeda, C.L.; Homma, S.; Chen, J.C.; Beermann, M.L.; Yan, C.; Emerson, C.P., Jr.; Miller, J.B.; Wagner, K.R.; et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenet. 2015, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Bosnakovski, D.; Toso, E.A.; Hartweck, L.M.; Magli, A.; Lee, H.A.; Thompson, E.R.; Dandapat, A.; Perlingeiro, R.C.R.; Kyba, M. The DUX4 homeodomains mediate inhibition of myogenesis and are functionally exchangeable with the Pax7 homeodomain. J. Cell Sci. 2017, 130, 3685–3697. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Himeda, C.L.; Jones, P.L. FSHD Therapeutic Strategies: What Will It Take to Get to Clinic? J. Pers. Med. 2022, 12, 865. https://doi.org/10.3390/jpm12060865
Himeda CL, Jones PL. FSHD Therapeutic Strategies: What Will It Take to Get to Clinic? Journal of Personalized Medicine. 2022; 12(6):865. https://doi.org/10.3390/jpm12060865
Chicago/Turabian StyleHimeda, Charis L., and Peter L. Jones. 2022. "FSHD Therapeutic Strategies: What Will It Take to Get to Clinic?" Journal of Personalized Medicine 12, no. 6: 865. https://doi.org/10.3390/jpm12060865
APA StyleHimeda, C. L., & Jones, P. L. (2022). FSHD Therapeutic Strategies: What Will It Take to Get to Clinic? Journal of Personalized Medicine, 12(6), 865. https://doi.org/10.3390/jpm12060865