
DNA Hypermethylation and a Specific Methylation Spectrum on the X Chromosome in Turner Syndrome as Determined by Nanopore Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Next Generation Sequencing
2.2. Nanopore Sequencing and Data Analysis
2.2.1. Sample Preparation
2.2.2. Library Construction and Sequencing
2.2.3. Bioinformatics Analysis
2.2.4. Methylation Analysis
2.2.5. Functional Enrichment Analysis
3. Results
3.1. Clinical Information
3.2. Origin of the X Chromosome and Casual Variants Analysis in TS
3.3. Genome-Wide Methylation Analysis
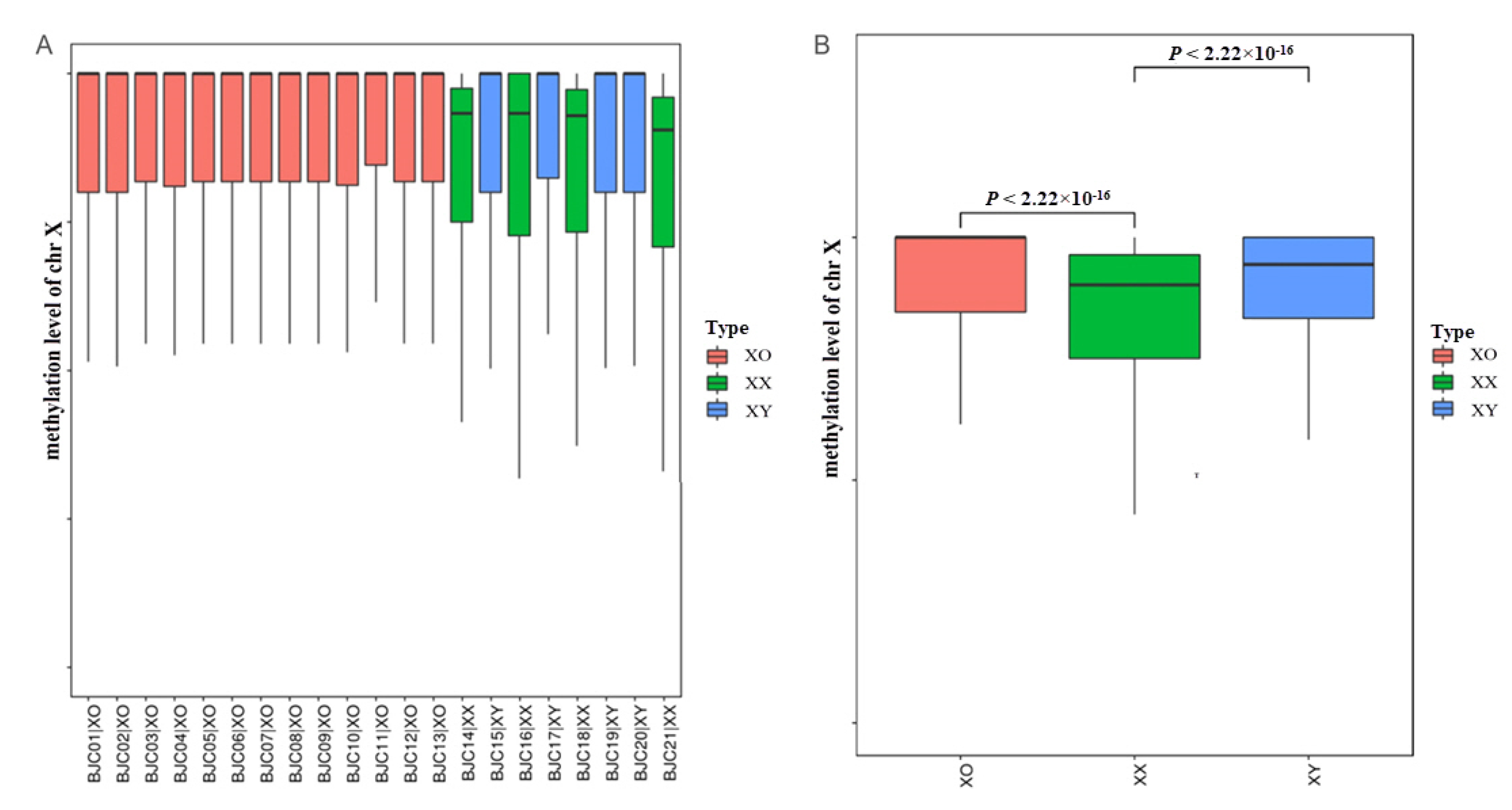
3.3.1. Overview of Genome-Wide Methylation in TS
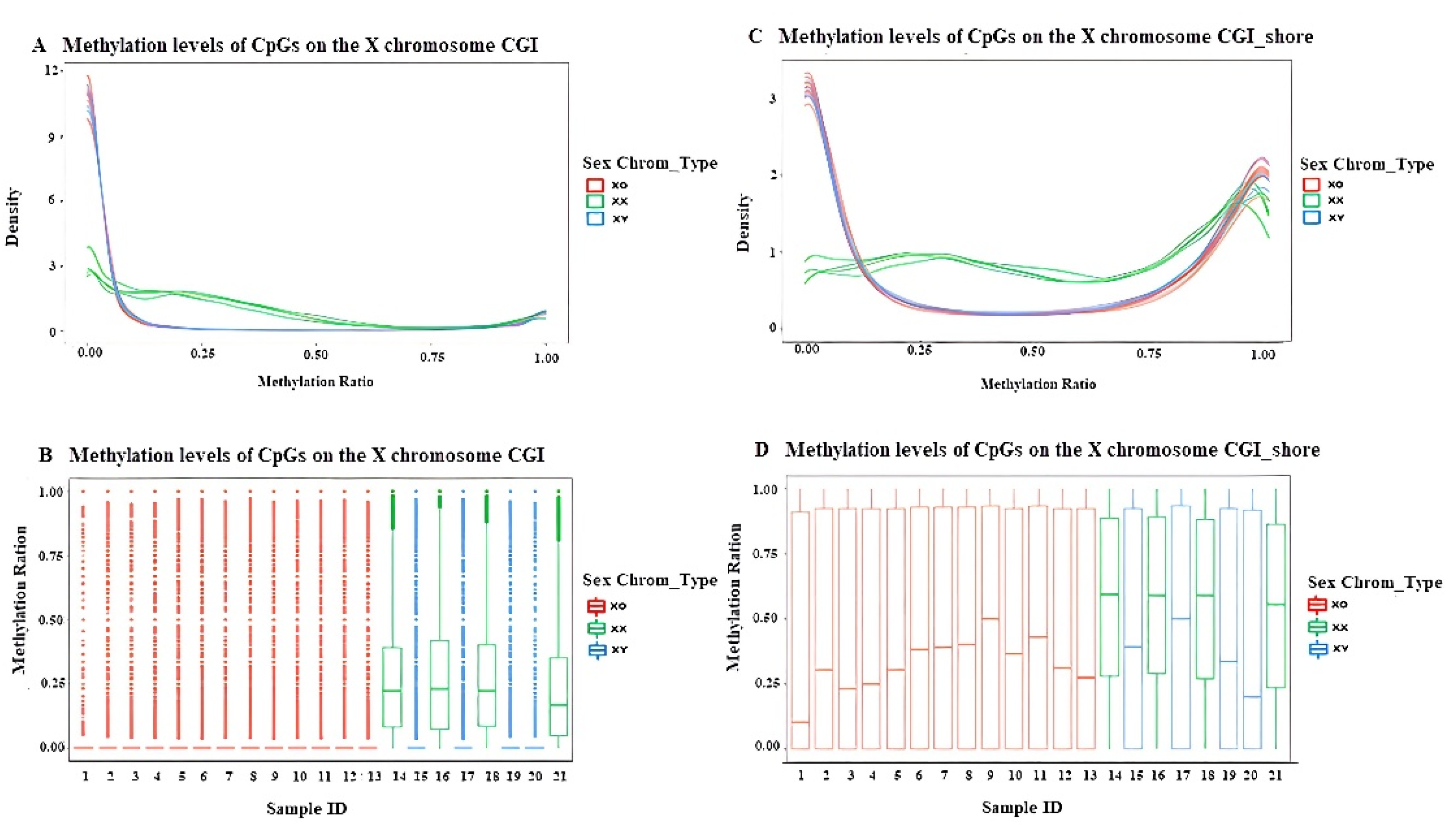
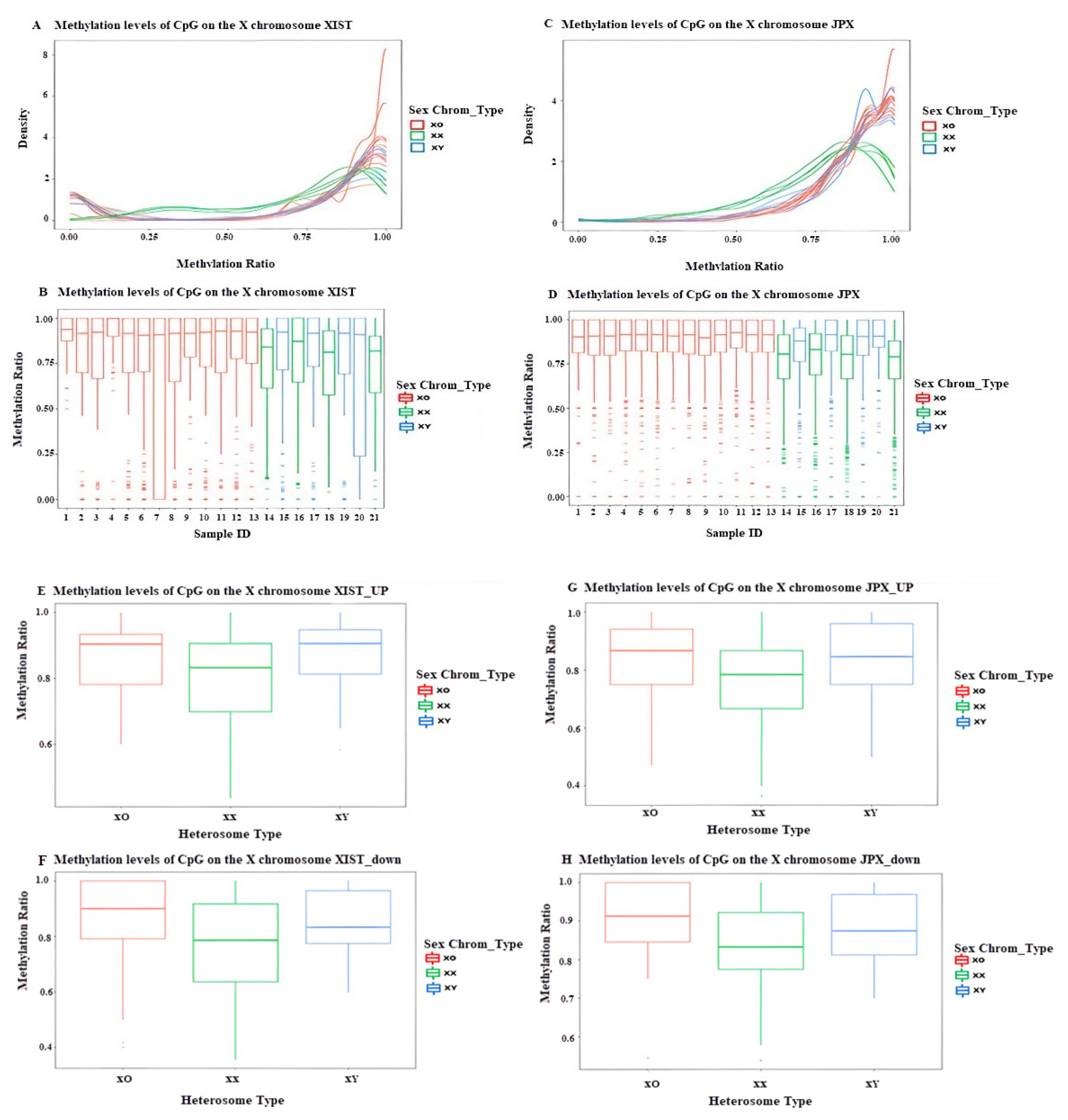
3.3.2. DNA Methylation Profiling of Different Functional Regions on the X Chromosome in TS
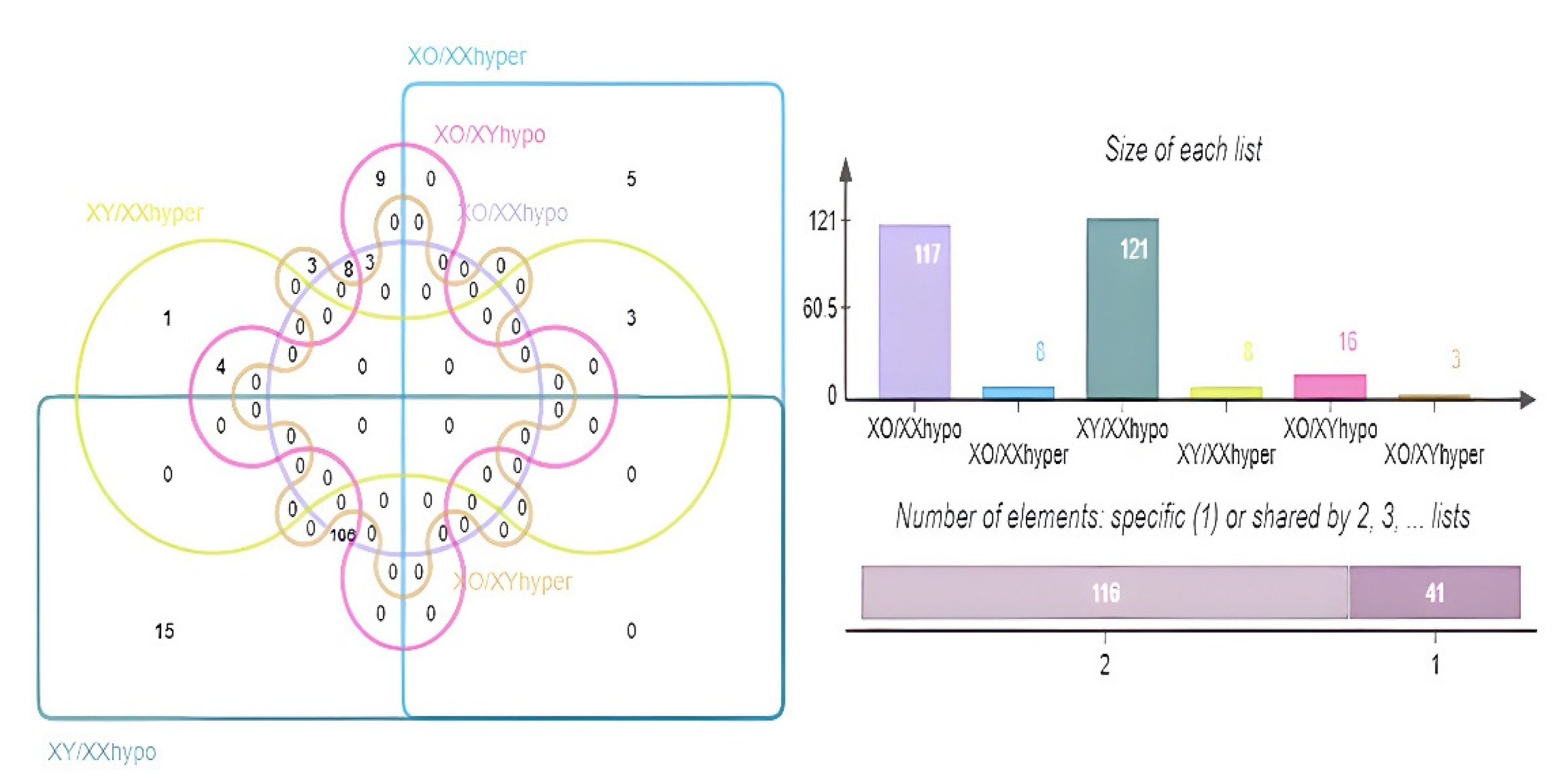
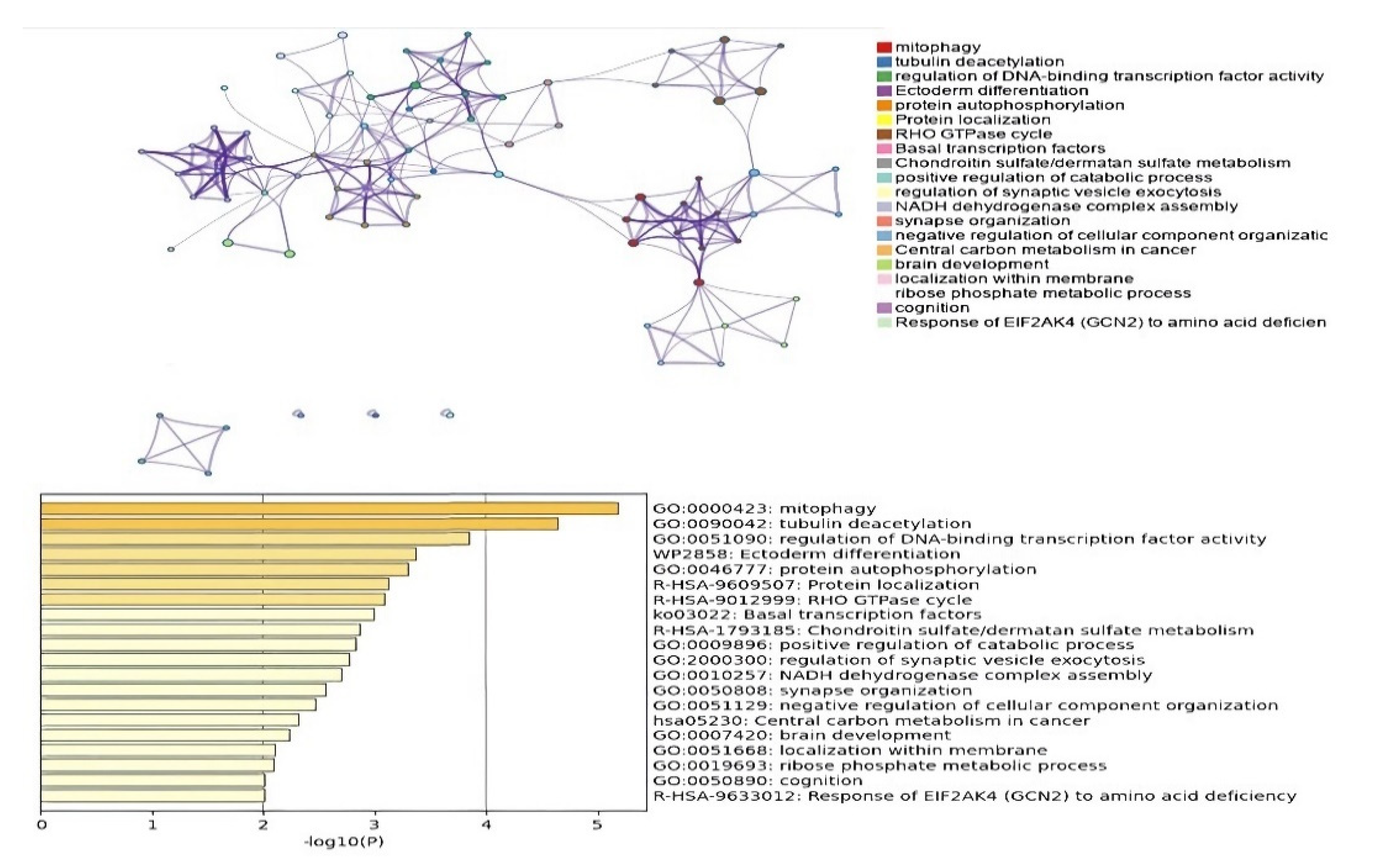
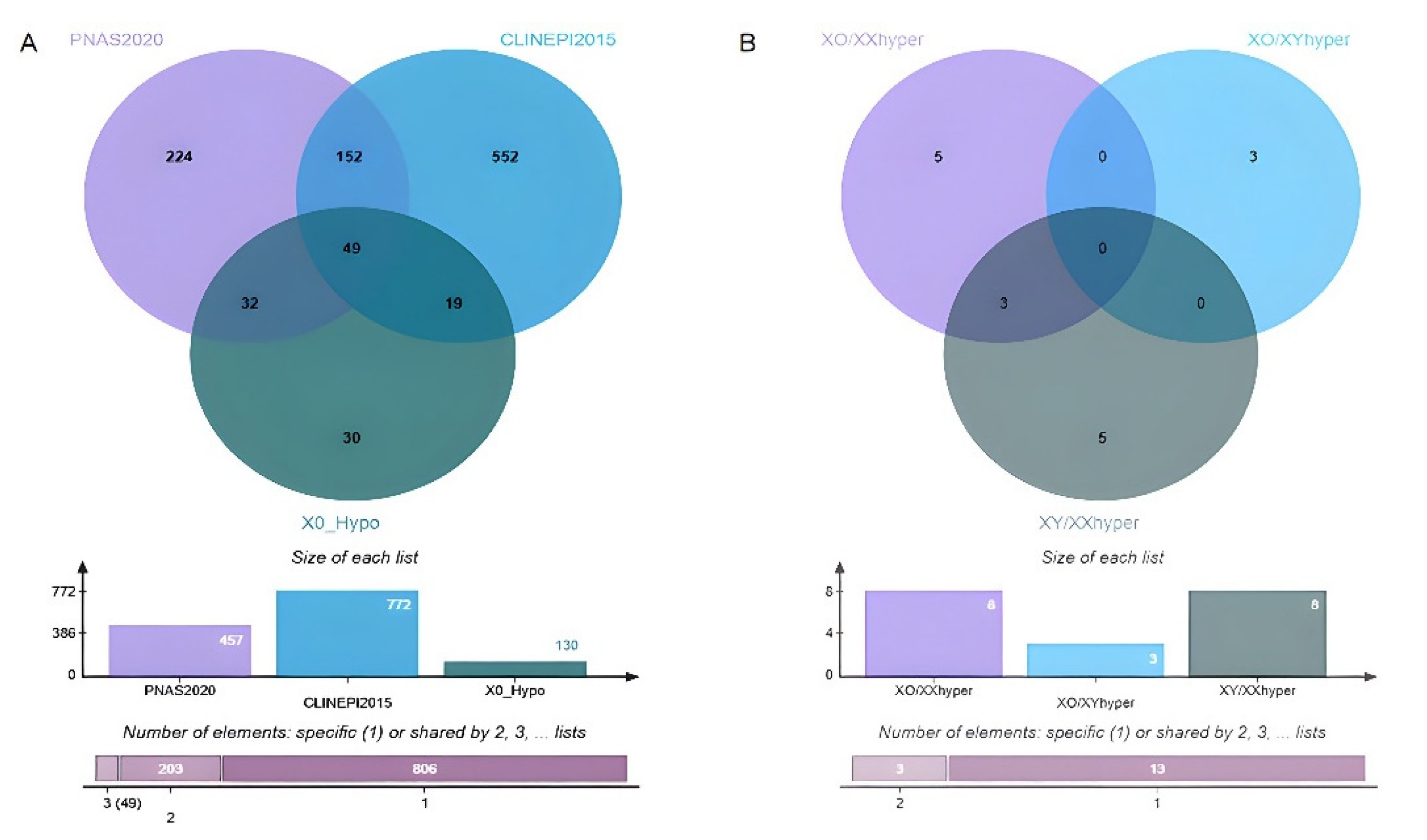
3.3.3. DMGs and Functional Enrichment Analysis
3.3.4. Potential Candidate Genes Associated with TS Phenotypes
4. Discussion
4.1. Genetic Information from Trio-WES
4.2. Profiling Genome-Wide and chrX DNA Methylation in TS
4.2.1. Genome-Wide DNA Methylation in TS
4.2.2. Profiling DNA Methylation on the X Chromosome
4.3. Differentially Methylated Genes (DMGs) and Functional Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvarez-Nava, F.; Lanes, R. Epigenetics in Turner syndrome. Clin. Epigenetics 2018, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Hook, E.B.; Warburton, D. Turner syndrome revisited: Review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014, 133, 417–424. [Google Scholar] [CrossRef]
- Zhang, X.; Hong, D.; Ma, S.; Ward, T.; Ho, M.; Pattni, R.; Duren, Z.; Stankov, A.; Shrestha, S.B.; Hallmayer, J.; et al. Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc. Natl. Acad. Sci. USA 2020, 117, 4864–4873. [Google Scholar] [CrossRef] [Green Version]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA methylation data by sequencing: Experimental approaches and recommendations for tools and pipelines for data analysis. Clin. Epigenetics 2019, 11, 193. [Google Scholar] [CrossRef] [Green Version]
- Rajpathak, S.N.; Deobagkar, D.D. Evidence for epigenetic alterations in Turner syndrome opens up feasibility of new pharmaceutical interventions. Curr. Pharm. Des. 2014, 20, 1778–1785. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.-L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10, 2449. [Google Scholar] [CrossRef] [Green Version]
- Locksley, R.M.; Nelson, C.S.; Fankhauser, J.E.; Klebanoff, S.J. Loss of granule myeloperoxidase during in vitro culture of human monocytes correlates with decay in antiprotozoa activity. Am. J. Trop. Med. Hyg. 1987, 36, 541–548. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The Long Noncoding RNA, Jpx, Is a Molecular switch for X chromosome inactivation. Cell 2010, 143, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Jamil, M.A.; Nuesgen, N.; Schreiner, F.; Priebe, L.; Hoffmann, P.; Herns, S.; Nöthen, M.M.; Fröhlich, H.; Oldenburg, J.; et al. DNA methylation signature in peripheral blood reveals distinct characteristics of human X chromosome numerical aberrations. Clin. Epigenetics 2015, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.L.; McNamara, E.A.; Longoni, M.; Miller, D.E.; Rohanizadegan, M.; Newman, L.A.; Hayes, F.; Levitsky, L.L.; Herrington, B.L.; Lin, A.E. Dual diagnoses in 152 patients with Turner syndrome: Knowledge of the second condition may lead to modification of treatment and/or surveillance. Am. J. Med. Genet. Part A 2018, 176, 2435–2445. [Google Scholar] [CrossRef]
- Li, L.; Li, Q.; Wang, Q.; Liu, L.; Li, R.; Liu, H.; He, Y.; Lash, G.E. Rare copy number variants in the genome of Chinese female children and adolescents with Turner syndrome. Biosci. Rep. 2019, 39, BSR20181305. [Google Scholar] [CrossRef] [Green Version]
- De Marqui, A.B.T. Turner syndrome and genetic polymorphism: A systematic review. Rev. Paul. Pediatr. 2015, 33, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, R.; Shukla, R.; Kabra, M.; Gupta, Y.; Jyotsna, V.P.; Khadgawat, R. Impact of parental origin of X-chromosome on clinical and biochemical profile in Turner syndrome. J. Pediatr. Endocrinol. Metab. 2020, 33, 1155–1163. [Google Scholar] [CrossRef]
- Hannon, E.; Knox, O.; Sugden, K.; Burrage, J.; Wong, C.C.Y.; Belsky, D.W.; Corcoran, D.L.; Arseneault, L.; Moffitt, T.E.; Caspi, A.; et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018, 14, e1007544. [Google Scholar] [CrossRef]
- Thunström, S.; Landin-Wilhelmsen, K.; Bryman, I.; Hanson, C. Side differences in the degree of mosaicism of the buccal mucosa in Turner syndrome. Mol. Genet. Genom. Med. 2019, 7, e00938. [Google Scholar] [CrossRef] [Green Version]
- Denes, A.-M.; Landin-Wilhelmsen, K.; Wettergren, Y.; Bryman, I.; Hanson, C. The proportion of diploid 46,XX cells increases with time in women with Turner syndrome—A 10-year follow-up study. Genet. Test. Mol. Biomark. 2015, 19, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Antequera, F. Structure, function and evolution of CpG island promoters. Cell Mol. Life Sci. 2003, 60, 1647–1658. [Google Scholar] [CrossRef]
- Trolle, C.; Nielsen, M.M.; Skakkebæk, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Ørntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220. [Google Scholar] [CrossRef] [Green Version]
- Schatz, M.C. Nanopore sequencing meets epigenetics. Nat. Methods 2017, 14, 347–348. [Google Scholar] [CrossRef]
- Guffanti, G.; Bartlett, A.; DeCrescenzo, P.; Macciardi, F.; Hunter, R. Transposable Elements. Curr. Top Behav. Neurosci. 2019, 42, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-E.; Ayarpadikannan, S.; Kim, H.-S. Role of transposable elements in genomic rearrangement, evolution, gene regulation and epigenetics in primates. Genes Genet. Syst. 2015, 90, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol. Spectr. 2015, 3, 1165–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Tolmacheva, E.N.; Vasilyev, S.A.; Lebedev, I.N. Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development. Genes 2020, 11, 1084. [Google Scholar] [CrossRef]
- Vasilyev, S.A.; Tolmacheva, E.N.; Vasilyeva, O.Y.; Markov, A.V.; Zhigalina, D.I.; Zatula, L.A.; Lee, V.A.; Serdyukova, E.S.; Sazhenova, E.A.; Nikitina, T.V.; et al. LINE-1 retrotransposon methylation in chorionic villi of first trimester miscarriages with aneuploidy. J. Assist. Reprod. Genet. 2021, 38, 139–149. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Huynh, K.D.; Fischle, W.; Verdin, E.; Bardwell, V.J. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000, 14, 1810–1823. [Google Scholar] [CrossRef]
- Fan, Z.; Yamaza, T.; Lee, J.S.; Yu, J.; Wang, S.; Fan, G.; Shi, S.; Wang, C.-Y. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell Biol. 2009, 11, 1002–1009. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Zhang, B.; Fan, L.; Chen, J.; Su, C.; Cao, B.; Wei, L.; Qin, M.; Gong, C. DNA Hypermethylation and a Specific Methylation Spectrum on the X Chromosome in Turner Syndrome as Determined by Nanopore Sequencing. J. Pers. Med. 2022, 12, 872. https://doi.org/10.3390/jpm12060872
Fan X, Zhang B, Fan L, Chen J, Su C, Cao B, Wei L, Qin M, Gong C. DNA Hypermethylation and a Specific Methylation Spectrum on the X Chromosome in Turner Syndrome as Determined by Nanopore Sequencing. Journal of Personalized Medicine. 2022; 12(6):872. https://doi.org/10.3390/jpm12060872
Chicago/Turabian StyleFan, Xin, Beibei Zhang, Lijun Fan, Jiajia Chen, Chang Su, Bingyan Cao, Liya Wei, Miao Qin, and Chunxiu Gong. 2022. "DNA Hypermethylation and a Specific Methylation Spectrum on the X Chromosome in Turner Syndrome as Determined by Nanopore Sequencing" Journal of Personalized Medicine 12, no. 6: 872. https://doi.org/10.3390/jpm12060872
APA StyleFan, X., Zhang, B., Fan, L., Chen, J., Su, C., Cao, B., Wei, L., Qin, M., & Gong, C. (2022). DNA Hypermethylation and a Specific Methylation Spectrum on the X Chromosome in Turner Syndrome as Determined by Nanopore Sequencing. Journal of Personalized Medicine, 12(6), 872. https://doi.org/10.3390/jpm12060872