
Additive Potentiation of R334W-CFTR Function by Novel Small Molecules

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Compounds
2.2. Generation of a New Cell Line
2.3. Cell Culture
2.4. HS-YFP Assay on a Plate Reader
2.5. In Silico Absorption, Distribution, Metabolism, and Excretion (ADME) Analyses
2.6. WB Analyses
2.7. Micro-Ussing Chamber Measurements
2.8. Fsk-Induced Swelling (FIS) Assay of Intestinal Organoids
2.9. Statistical Analyses
3. Results
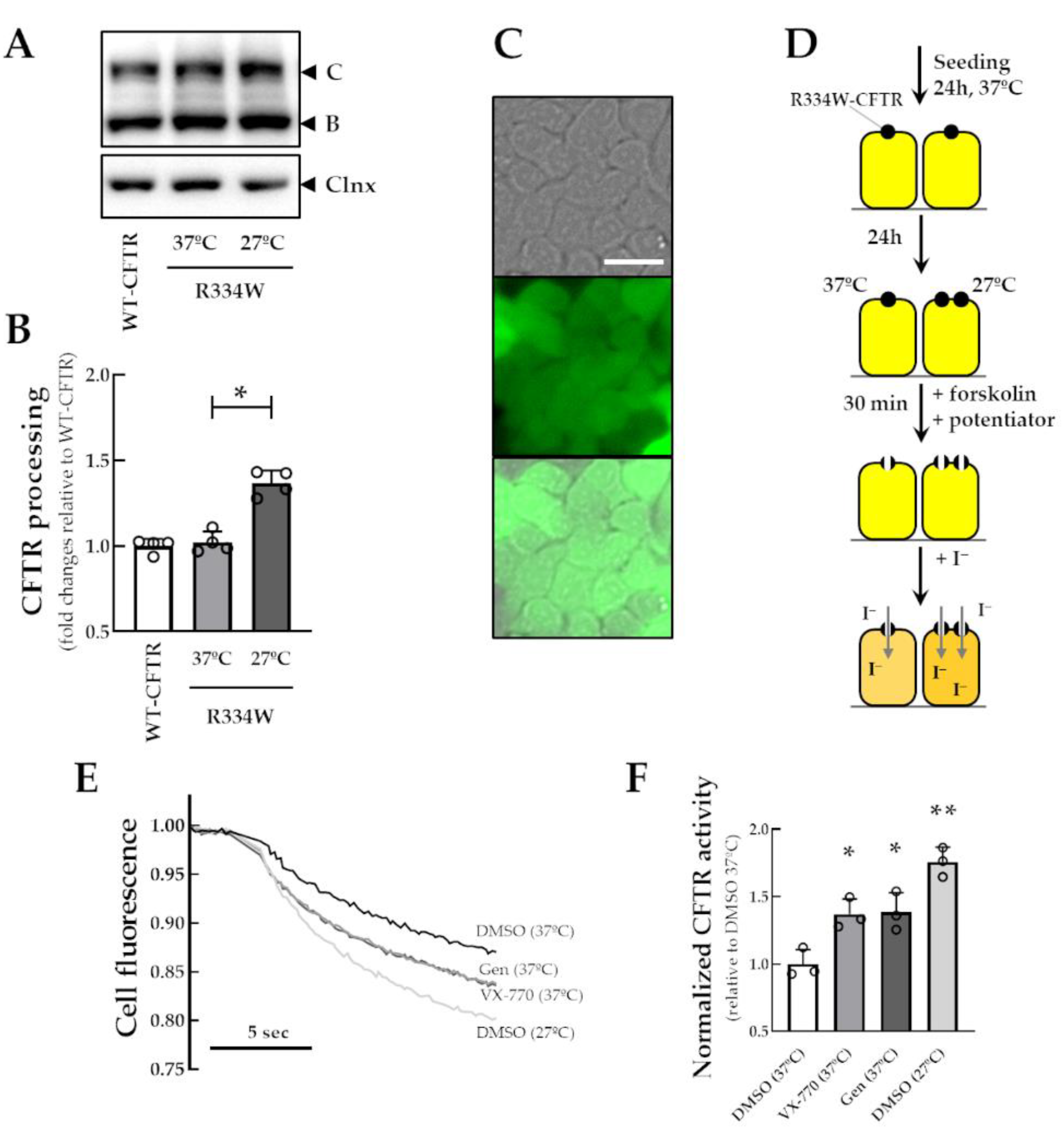
3.1. Characterization of a New Cell Line Co-Expressing the HS-YFP and R334W-CFTR
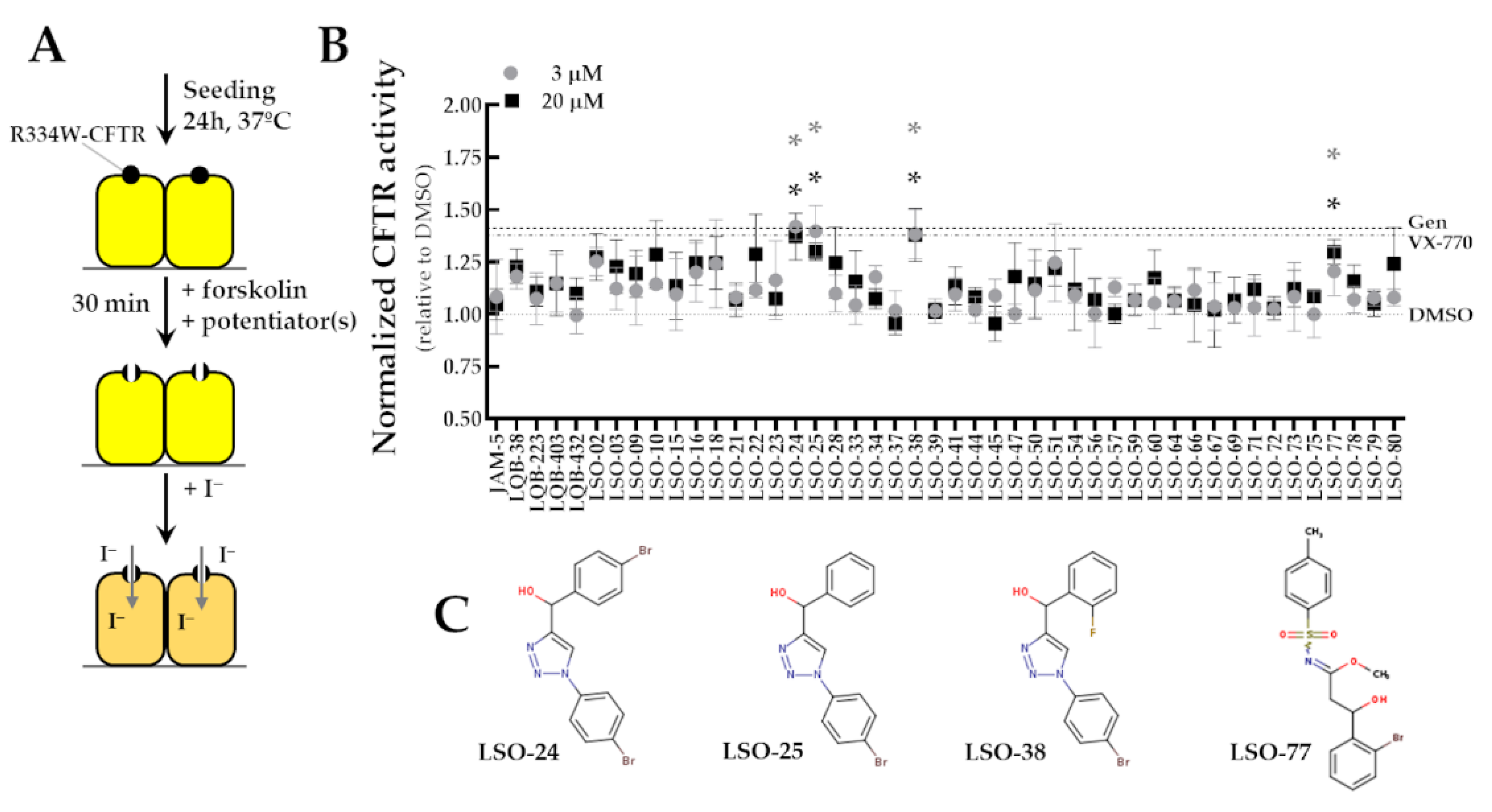
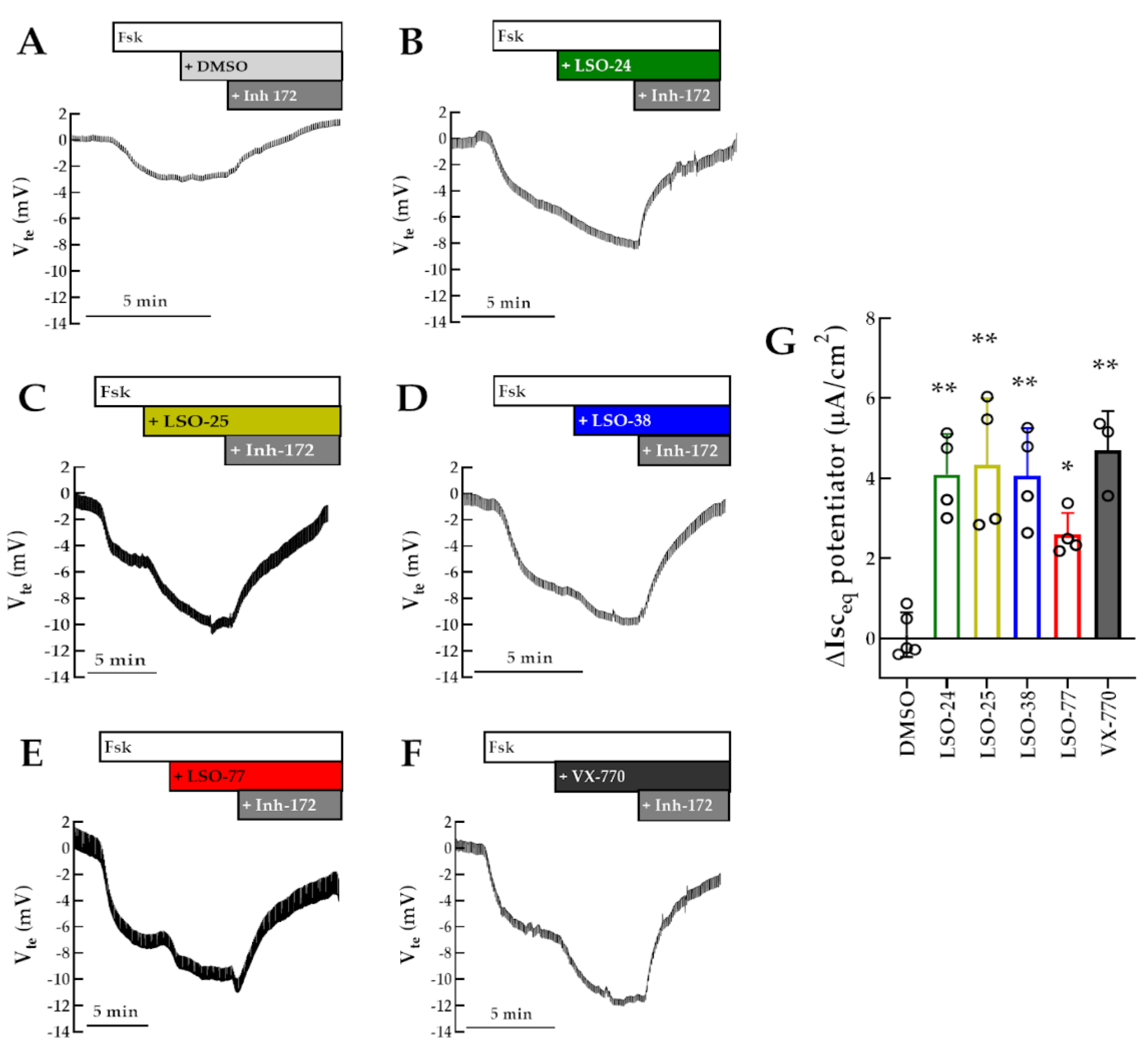
3.2. Identification of Novel Potentiators for the Rescue of R334W-CFTR Function
3.3. Assessment of Potentiator Activity for WT-CFTR
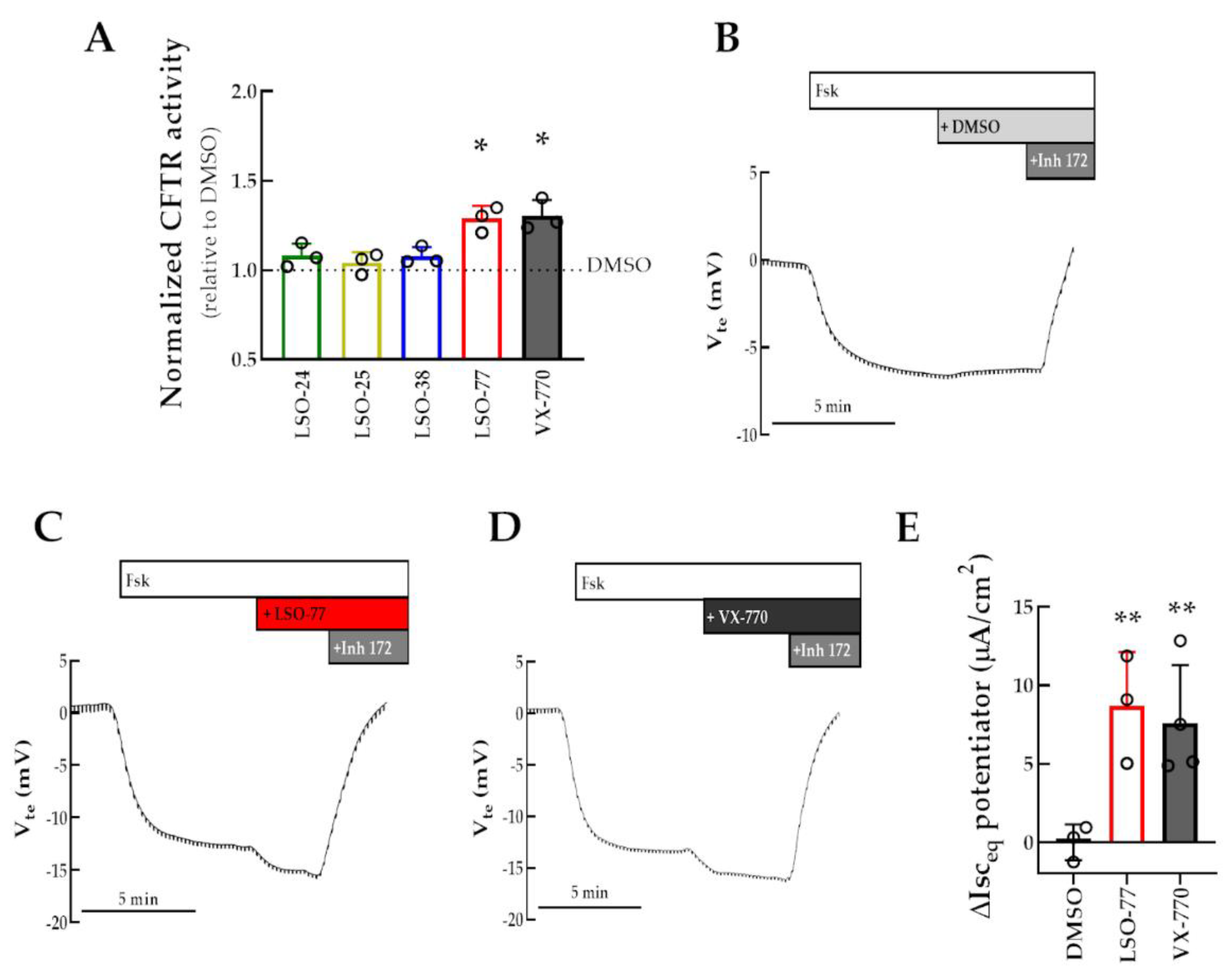
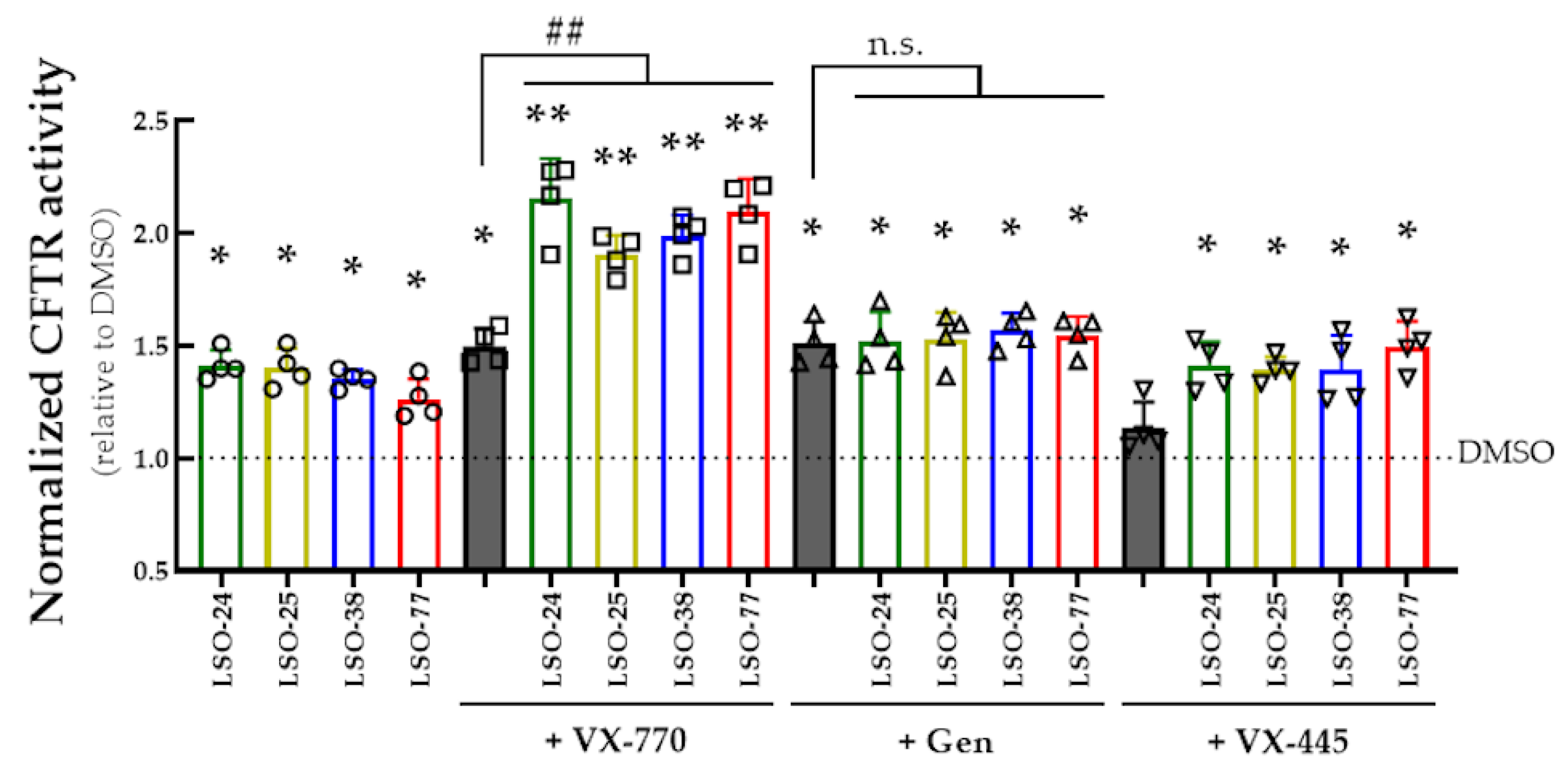
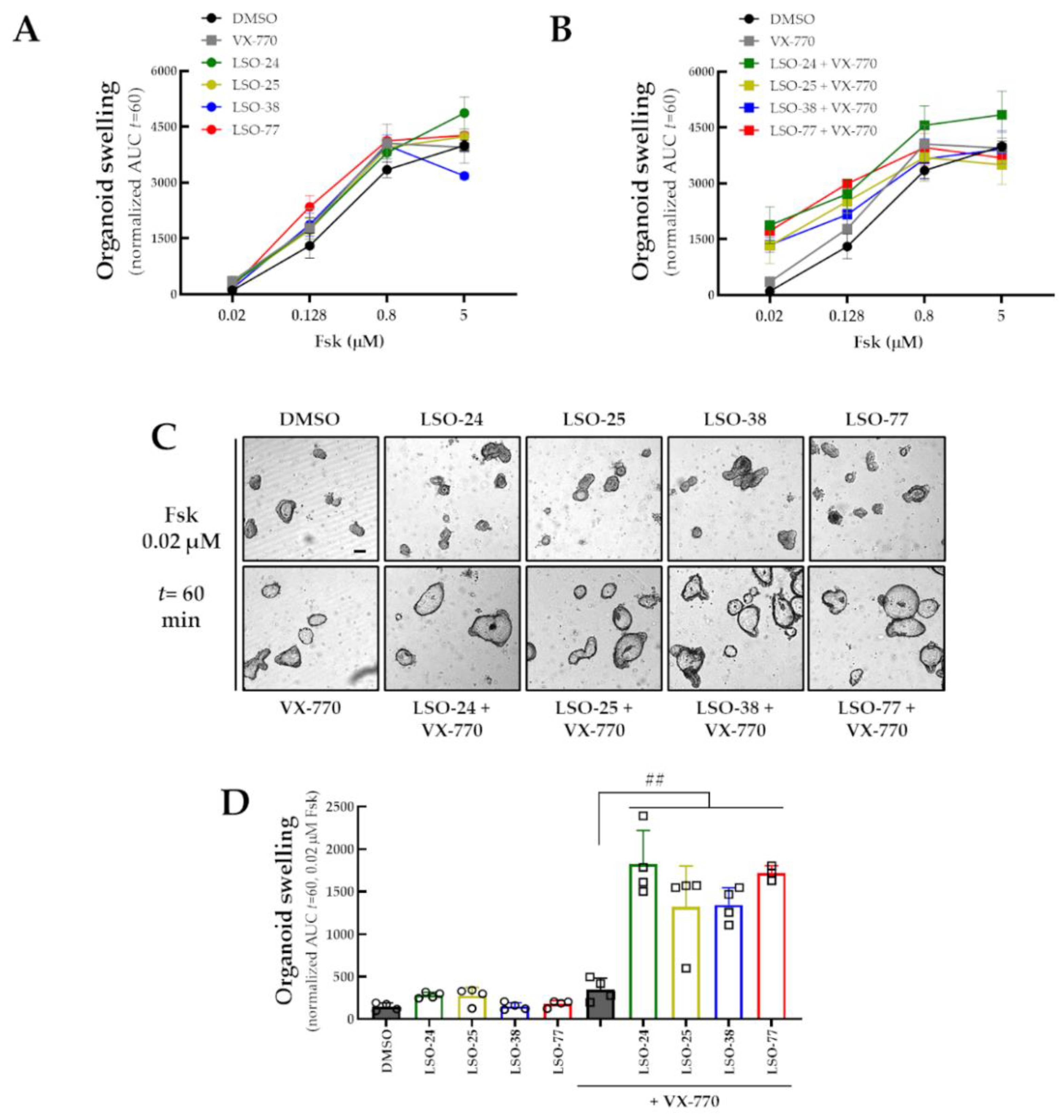
3.4. Assessment of Co-Potentiator Activity for R334W-CFTR in a Cell Line and in Intestinal Organoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- Eckford, P.D.W.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessonova, L.; Volkova, N.; Higgins, M.; Bengtsson, L.; Tian, S.; Simard, C.; Konstan, M.W.; Sawicki, G.S.; Sewall, A.; Nyangoma, S.; et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018, 73, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- ECFS Patient Registry Annual Report 2020. Available online: https://www.ecfs.eu/sites/default/files/ECFSPR_Report_2020_v1.0%20%2807Jun2022%29_website.pdf (accessed on 10 October 2022).
- Relatório do Registro Brasileiro de Fibrose Cística 2019. Available online: http://portalgbefc.org.br/ckfinder/userfiles/files/REBRAFC_2019.pdf (accessed on 10 October 2022).
- Sheppard, D.N.; Rich, D.P.; Ostedgaard, L.S.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Mutations in CFTR associated with mild-disease-form CI- channels with altered pore properties. Nature 1993, 362, 160–164. [Google Scholar] [CrossRef]
- Smith, S.S.; Liu, X.; Zhang, Z.R.; Sun, F.; Kriewall, T.E.; Mccarty, N.A.; Dawson, D.C. CFTR: Covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 2001, 118, 407–431. [Google Scholar] [CrossRef]
- Phuan, P.W.; Tan, J.A.; Rivera, A.A.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Haggie, P.M.; Verkman, A.S. Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J. Cyst. Fibros. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Jih, K.Y.; Hwang, T.C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef] [Green Version]
- Phuan, P.W.; Son, J.H.; Tan, J.A.; Li, C.; Musante, I.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Kurth, M.J.; Galietta, L.J.; et al. Combination potentiator (‘co-potentiator’) therapy for CF caused by CFTR mutants, including N1303K, that are poorly responsive to single potentiators. J. Cyst. Fibros. 2018, 17, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. 2021, 20, 895–898. [Google Scholar] [CrossRef]
- Gees, M.; Musch, S.; Van Der Plas, S.; Wesse, A.S.; Vandevelde, A.; Verdonck, K.; Mammoliti, O.; Hwang, T.C.; Sonck, K.; Stouten, P.; et al. Identification and characterization of novel CFTR potentiators. Front. Pharmacol. 2018, 9, 1221. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Berg, A.P.; Wang, Y.; Jantarajit, W.; Sutcliffe, K.J.; Stevens, E.B.; Cao, L.; Pregel, M.J.; Sheppard, D.N. A small molecule CFTR potentiator restores ATP-dependent channel gating to the cystic fibrosis mutant G551D-CFTR. Br. J. Pharmacol. 2022, 179, 1319–1337. [Google Scholar] [CrossRef]
- Moran, O.; Galietta, L.J.V.; Zegarra-Moran, O. Binding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domains. Cell. Mol. Life Sci. 2005, 62, 446–460. [Google Scholar] [CrossRef]
- Wang, X.; Huang, B.; Liu, X.; Zhan, P. Discovery of bioactive molecules from CuAAC click-chemistry-based combinatorial libraries. Drug Discov. Today 2016, 21, 118–132. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Kant, R.; Kumar, D.; Agarwal, D.; Gupta, R.D.; Tilak, R.; Awasthi, S.K.; Agarwal, A. Synthesis of newer 1,2,3-triazole linked chalcone and flavone hybrid compounds and evaluation of their antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2016, 113, 34–49. [Google Scholar] [CrossRef]
- Kim, T.W.; Yong, Y.; Shin, S.Y.; Jung, H.; Park, K.H.; Lee, Y.H.; Lim, Y.; Jung, K.Y. Synthesis and biological evaluation of phenyl-1H-1,2,3-triazole derivatives as anti-inflammatory agents. Bioorg. Chem. 2015, 59, 1–11. [Google Scholar] [CrossRef]
- Ferreira, M.D.L.G.; Pinheiro, L.C.S.; Santos-Filho, O.A.; Peçanha, M.D.S.; Sacramento, C.Q.; Machado, V.; Ferreira, V.F.; Souza, T.M.L.; Boechat, N. Design, synthesis, and antiviral activity of new 1H-1,2,3-triazole nucleoside ribavirin analogs. Med. Chem. Res. 2014, 23, 1501–1511. [Google Scholar] [CrossRef]
- Stefely, J.A.; Palchaudhuri, R.; Miller, P.A.; Peterson, R.J.; Moraski, G.C.; Hergenrother, P.J.; Miller, M.J. N -((1-benzyl-1 H -1,2,3-triazol-4-yl)methyl)arylamide as a new scaffold that provides rapid access to antimicrotubule agents: Synthesis and evaluation of antiproliferative activity against select cancer cell lines. J. Med. Chem. 2010, 53, 3389–3395. [Google Scholar] [CrossRef] [Green Version]
- Naaz, F.; Preeti Pallavi, M.C.; Shafi, S.; Mulakayala, N.; Shahar Yar, M.; Sampath Kumar, H.M. 1,2,3-triazole tethered Indole-3-glyoxamide derivatives as multiple inhibitors of 5-LOX, COX-2 & tubulin: Their anti-proliferative & anti-inflammatory activity. Bioorg. Chem. 2018, 81, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, V.D.; de Faria, B.M.; Colombo, E.; Ascari, L.; Freitas, G.P.A.; Flores, L.S.; Cordeiro, Y.; Romão, L.; Buarque, C.D. Design, synthesis, structural characterization and in vitro evaluation of new 1,4-disubstituted-1,2,3-triazole derivatives against glioblastoma cells. Bioorg. Chem. 2019, 83, 87–97. [Google Scholar] [CrossRef]
- Da Silva, V.D.; Silva, R.R.; Gonçalves Neto, J.; López-Corcuera, B.; Guimarães, M.Z.; Noël, F.; Buarque, C.D. New α-Hydroxy-1,2,3-triazoles and 9H-Fluorenes-1,2,3-triazolesSynthesis and Evaluation as Glycine Transporter 1 Inhibitors. J. Braz. Chem. Soc. 2020, 31, 1258–1269. [Google Scholar] [CrossRef]
- Bacalhau, M.; Ferreira, F.C.; Kmit, A.; Souza, F.R.; da Silva, V.D.; Pimentel, A.S.; Amaral, M.D.; Buarque, C.D.; Lopes-Pacheco, M. Identification of novel F508del-CFTR traffic correctors among triazole derivatives. Eur. J. Pharmacol. 2023, 938, 175396. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Bebok, Z.; Collawn, J.F.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.J.; Clancy, J.P. Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef]
- Pedemonte, N.; Zegarra-moran, O.; Galietta, L.J. V High-throughput screening of libraries of compounds to identify CFTR modulators. Methods Mol. Biol. 2011, 741, 13–21. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M.; Silva, I.A.L.; Turner, M.J.; Carlile, G.W.; Sondo, E.; Thomas, D.Y.; Pedemonte, N.; Hanrahan, J.W.; Amaral, M.D. Characterization of the mechanism of action of RDR01752, a novel corrector of F508del-CFTR. Biochem. Pharmacol. 2020, 180, 114133. [Google Scholar] [CrossRef]
- Awatade, N.T.; Uliyakina, I.; Farinha, C.M.; Clarke, L.A.; Mendes, K.; Solé, A.; Pastor, J.; Ramos, M.M.; Amaral, M.D. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine 2015, 2, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Varga, K.; Goldstein, R.F.; Jurkuvenaite, A.; Chen, L.; Matalon, S.; Sorscher, E.J.; Bebok, Z.; Collawn, J.F. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 2008, 410, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Combination of Correctors Rescue ΔF508-CFTR by Reducing Its Association with Hsp40 and Hsp27. J. Biol. Chem. 2015, 290, 25636–25645. [Google Scholar] [CrossRef] [Green Version]
- Van Der Plas, S.E.; Kelgtermans, H.; Mammoliti, O.; Menet, C.; Tricarico, G.; De Blieck, A.; Joannesse, C.; De Munck, T.; Lambin, D.; Cowart, M.; et al. Discovery of GLPG2451, a Novel Once Daily Potentiator for the Treatment of Cystic Fibrosis. J. Med. Chem. 2021, 64, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-I.; Qiu, L.; Sohma, Y.; Conrath, K.; Zou, X.; Hwang, T.-C. Identifying the molecular target sites for CFTR potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019, 151, 912–928. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.-I.; Sohma, Y.; Conrath, K.; Hwang, T.-C. A common mechanism for CFTR potentiators. J. Gen. Physiol. 2017, 149, 1105–1118. [Google Scholar] [CrossRef] [Green Version]
- Doiron, J.E.; Le, C.A.; Ody, B.K.; Brace, J.B.; Post, S.J.; Thacker, N.L.; Hill, H.M.; Breton, G.W.; Mulder, M.J.; Chang, S.; et al. Evaluation of 1,2,3-Triazoles as Amide Bioisosteres In Cystic Fibrosis Transmembrane Conductance Regulator Modulators VX-770 and VX-809. Chemistry 2019, 25, 3662–3674. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Cuyx, S.; Van Biervliet, S.; Dupont, L.; Christ, F.; Debyser, Z.; Vermeulen, F.; Carlon, M.S. Novel CFTR modulator combinations maximise rescue of G85E and N1303K in rectal organoids. ERJ Open Res. 2022, 8, 00716–02021. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Wainwright, C.E.; Elizabeth Tullis, D.; Rodriguez, S.; Niknian, M.; Higgins, M.; Davies, J.C.; Wagener, J.S. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D-CFTR mutation treated with ivacaftor. J. Cyst. Fibros. 2018, 17, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef]
- Van Willigen, M.; Vonk, A.M.; Yeoh, H.Y.; Kruisselbrink, E.; Kleizen, B.; Van Der Ent, C.K.; Egmond, M.R.; De Jonge, H.R.; Braakman, I.; Beekman, J.M.; et al. Folding-function relationship of the most common cystic fibrosis-causing CFTR conductance mutants. Life Sci. Alliance 2019, 2, 2201800172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitropoulou, G.; Brandenberg, N.; Hoehnel, S.; Ceroni, C.; Balmpouzis, Z.; Blanchon, S.; Dorta, G.; Sauty, A.; Koutsokera, A. Rectal organoid-guided CFTR modulator therapy restores lung function in a CF patient with the rare 1677delTA/R334W genotype. Eur. Respir. J. 2022, 60, 2201341. [Google Scholar] [CrossRef]
- Ciciriello, F.; Bijvelds, M.J.C.; Alghisi, F.; Meijsen, K.F.; Cristiani, L.; Sorio, C.; Melotti, P.; Fiocchi, A.G.; Lucidi, V.; De Jonge, H.R. Theratyping of the Rare CFTR Variants E193K and R334W in Rectal Organoid-Derived Epithelial Monolayers. J. Pers. Med. 2022, 12, 632. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (µM) | Emax (µM) |
|---|---|---|
| LSO-24 | 1.24 | 3.6 |
| LSO-25 | 1.41 | 4.3 |
| LSO-38 | 1.81 | 3.7 |
| LSO-77 | 2.96 | 6.9 |
| Comp | MW | nHBD | nHBA | nRB | LogP | TPSA (Å) | GI Absorption | PAINS Alert |
|---|---|---|---|---|---|---|---|---|
| LSO-24 | 409.08 | 1 | 3 | 3 | 3.54 | 50.94 | High | 0 |
| LSO-25 | 330.18 | 1 | 3 | 3 | 2.77 | 50.94 | High | 0 |
| LSO-38 | 348.17 | 1 | 4 | 3 | 3.11 | 50.94 | High | 0 |
| LSO-77 | 412.30 | 1 | 5 | 6 | 3.60 | 84.34 | High | 0 |
| VX-770 | 392.49 | 3 | 3 | 5 | 4.47 | 82.19 | High | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacalhau, M.; Ferreira, F.C.; Silva, I.A.L.; Buarque, C.D.; Amaral, M.D.; Lopes-Pacheco, M. Additive Potentiation of R334W-CFTR Function by Novel Small Molecules. J. Pers. Med. 2023, 13, 102. https://doi.org/10.3390/jpm13010102
Bacalhau M, Ferreira FC, Silva IAL, Buarque CD, Amaral MD, Lopes-Pacheco M. Additive Potentiation of R334W-CFTR Function by Novel Small Molecules. Journal of Personalized Medicine. 2023; 13(1):102. https://doi.org/10.3390/jpm13010102
Chicago/Turabian StyleBacalhau, Mafalda, Filipa C. Ferreira, Iris A. L. Silva, Camilla D. Buarque, Margarida D. Amaral, and Miquéias Lopes-Pacheco. 2023. "Additive Potentiation of R334W-CFTR Function by Novel Small Molecules" Journal of Personalized Medicine 13, no. 1: 102. https://doi.org/10.3390/jpm13010102
APA StyleBacalhau, M., Ferreira, F. C., Silva, I. A. L., Buarque, C. D., Amaral, M. D., & Lopes-Pacheco, M. (2023). Additive Potentiation of R334W-CFTR Function by Novel Small Molecules. Journal of Personalized Medicine, 13(1), 102. https://doi.org/10.3390/jpm13010102