

Exploring the Role of Biomarkers Associated with Alveolar Damage and Dysfunction in Idiopathic Pulmonary Fibrosis—A Systematic Review

 , ,
, , 
{kind=link}
Abstract
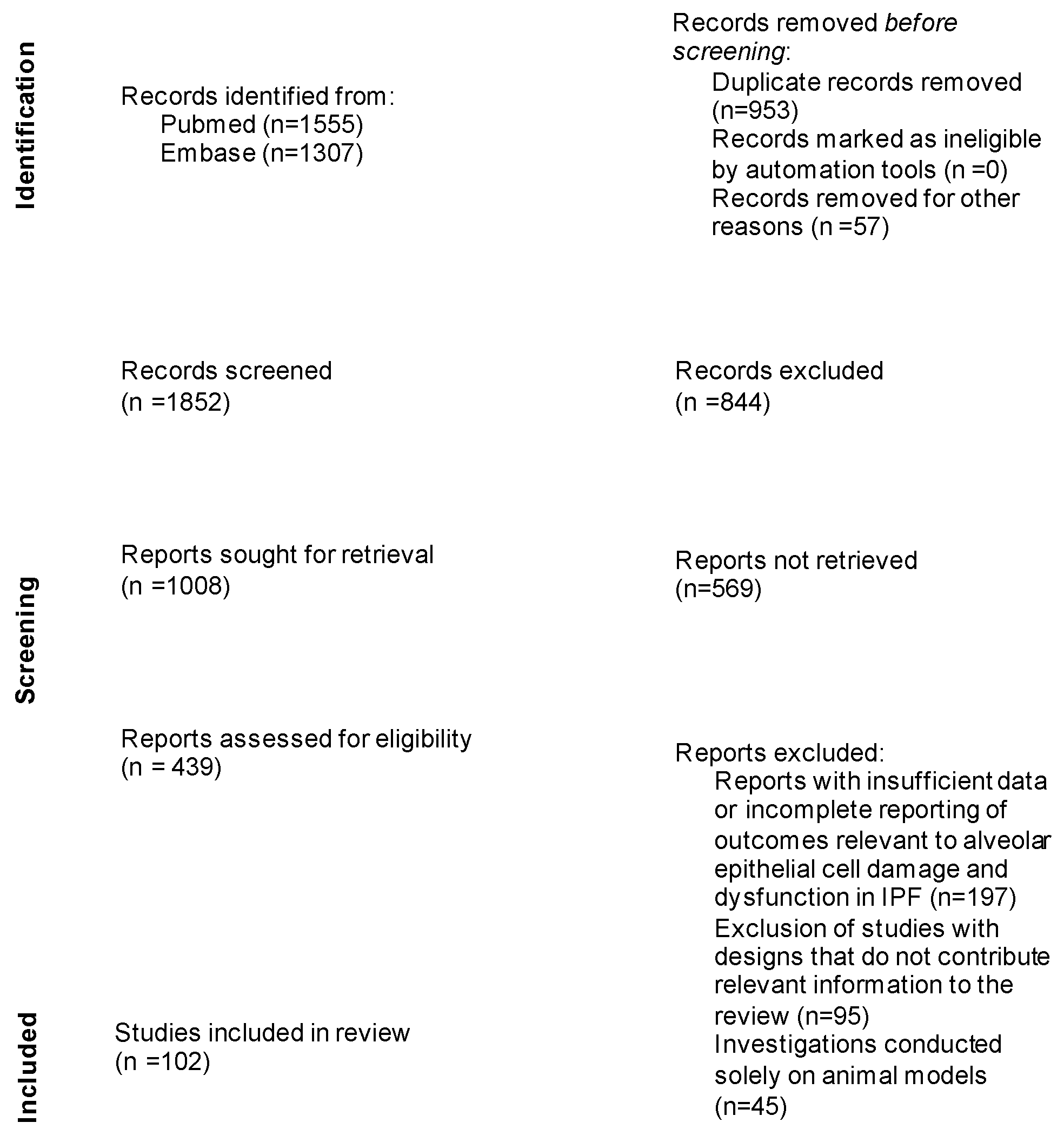
:1. Introduction
- Serum biomarkers, such as Krebs von den Lungen-6 (KL-6), surfactant protein D (SP-D), and serum matrix metalloproteinase 7 (MMP-7) have been shown to be useful in the diagnosis of IPF. However, their sensitivity and specificity are not high enough to be used as standalone diagnostic tools;
- Bronchoalveolar lavage fluid biomarkers, such as SP-D, KL-6, and MMP-7 have been found in bronchoalveolar lavage fluid samples from patients with IPF. Their diagnostic accuracy is limited by the fact that they are not specific to IPF and can be found in other lung diseases;
- Exhaled breath condensate biomarkers, such as hydrogen peroxide, nitric oxide, and leukotrienes have been studied in the context of IPF diagnosis. However, their diagnostic accuracy is limited by the fact that they are not specific to IPF and can be influenced by other factors such as smoking;
- Lung tissue biomarkers, similar to serum biomarkers, have been studied in the context of IPF diagnosis but their diagnostic accuracy is limited since confirming their presence in the lung tissue requires invasive procedures such as lung biopsy;
- Genetic biomarkers, such as mucin 5B (MUC5B) and telomerase reverse transcriptase (TERT) have been associated with an increased risk of developing IPF, but their diagnostic accuracy is limited by the fact that they are not specific to IPF and can also be found in healthy individuals;
- Proteomic biomarkers, such as serum amyloid (SAA) and α1 antitrypsin (AAT) seem to be useful in the IPF diagnosis, but their accuracy is limited by their non-specificity to IPF and can be found in other lung diseases;
- Metabolomic biomarkers, such as sphingolipids and glycerophospholipids have been evoked in the context of IPF diagnosis, but their values are highly biased by the influence of rather uncontrollable factors, such as diet;
2. Materials and Methods
3. Results
3.1. Biomarkers
3.2. Krebs von den Lungen 6 (KL-6)
3.3. Surfactant Proteins
3.4. The Mucin MUC5B
3.5. Oncomarkers (CA 15-3, CA 19-9, CA 125, and CEA)
3.5.1. CA 15-3
3.5.2. CA 19-9
3.5.3. CA 125
3.5.4. CEA
3.6. Clara Cell Secretory Protein (CC16)
3.7. Telomere Shortening
3.8. Cleaved Cytokeratin 18 (cCK-18)
3.9. Alpha-v beta-6 (αvβ6) Integrin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic Idiopathic Interstitial Lung Disease: The Molecular and Cellular Key Players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Ortega, P.; Molina-Molina, M. Interstitial Lung Diseases in Developing Countries. Ann. Glob. Health 2019, 85, 4. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Daniels, C.E.; Hartman, T.E.; Yi, E.S. Diagnosis of Interstitial Lung Diseases. Mayo Clin. Proc. 2007, 82, 976–986. [Google Scholar] [CrossRef]
- Maher, T.M.; Strek, M.E. Antifibrotic Therapy for Idiopathic Pulmonary Fibrosis: Time to Treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef]
- Munchel, J.K.; Shea, B.S. Diagnosis and Management of Idiopathic Pulmonary Fibrosis. Rhode Isl. Med. J. 2021, 104, 26–29. [Google Scholar]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; Kasselman, L.J.; Glass, A.D.; DeLeon, J.; Reiss, A.B. Idiopathic Pulmonary Fibrosis: Molecular Mechanisms and Potential Treatment Approaches. Respir. Investig. 2020, 58, 320–335. [Google Scholar] [CrossRef]
- Behr, J.; Günther, A.; Bonella, F.; Dinkel, J.; Fink, L.; Geiser, T.; Geissler, K.; Gläser, S.; Handzhiev, S.; Jonigk, D.; et al. S2K Guideline for Diagnosis of Idiopathic Pulmonary Fibrosis. Respiration 2021, 100, 238–271. [Google Scholar] [CrossRef]
- Patel, H.; Shah, J.R.; Patel, D.R.; Avanthika, C.; Jhaveri, S.; Gor, K. Idiopathic Pulmonary Fibrosis: Diagnosis, Biomarkers and Newer Treatment Protocols. Dis. Mon. 2023, 69, 101484. [Google Scholar] [CrossRef]
- Elhai, M.; Avouac, J.; Allanore, Y. Circulating Lung Biomarkers in Idiopathic Lung Fibrosis and Interstitial Lung Diseases Associated with Connective Tissue Diseases: Where Do We Stand? Semin. Arthritis Rheum. 2020, 50, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Hambly, N.; Shimbori, C.; Kolb, M. Molecular Classification of Idiopathic Pulmonary Fibrosis: Personalized Medicine, Genetics and Biomarkers. Respirology 2015, 20, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, S.; Stjepanovic, M.; Asanin, M. Biomarkers in Idiopathic Pulmonary Fibrosis. In Idiopathic Pulmonary Fibrosis; Surani, S., Rajasurya, V., Eds.; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Soccio, P.; Moriondo, G.; Scioscia, G.; Leo, V.; Tondo, P.; Salerno, L.; Palange, P.; Barbaro, M.P.F.; Lacedonia, D. Searching for airways biomarkers useful to identify progressive pulmonary fibrosis. BMC Pulm. Med. 2023, 23, 407. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.S.; Echt, G.A.; Oldham, J.M. Biomarkers in Progressive Fibrosing Interstitial Lung Disease: Optimizing Diagnosis, Prognosis, and Treatment Response. Front. Med. 2021, 8, 680997. [Google Scholar] [CrossRef]
- Todd, J.L.; Neely, M.L.; Overton, R.; Durham, K.; Gulati, M.; Huang, H.; Roman, J.; Newby, L.K.; Flaherty, K.R.; Vinisko, R.; et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir. Res. 2019, 20, 227. [Google Scholar] [CrossRef]
- Sood, S.; Russell, T.; Shifren, A. Biomarkers in Idiopathic Pulmonary Fibrosis. Respiratory Medicine; Humana Press: Cham, Switzerland, 2018; pp. 241–271. [Google Scholar] [CrossRef]
- Bodaghi, A.; Fattahi, N.; Ramazani, A. Biomarkers: Promising and Valuable Tools towards Diagnosis, Prognosis and Treatment of COVID-19 and Other Diseases. Heliyon 2023, 9, e13323. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker Definitions and Their Applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Vij, R.; Noth, I. Peripheral Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Transl. Res. 2012, 159, 218–227. [Google Scholar] [CrossRef]
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Wujak, L.; Wygrecka, M.; Markart, P. Biomarkers in Idiopathic Pulmonary Fibrosis. Matrix Biol. 2018, 68–69, 404–421. [Google Scholar] [CrossRef]
- Kolb, M.; Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers—Insights from the Bench Side. Respiration 2013, 86, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The α v β 1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Trans. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Ballester, B.; Montero, P.; Escriva, J.; Artigues, E.; Alós, M.; Pastor-Clerigues, A.; Morcillo, E.; Cortijo, J. MUC1 intracellular bioactivation mediates lung fibrosis. Thorax 2020, 75, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.G.; Choi, S.M.; Lee, J.H.; Yoon, J.-K.; Song, J.W. Changes in Blood Krebs von Den Lungen-6 Predict the Mortality of Patients with Acute Exacerbation of Interstitial Lung Disease. Sci. Rep. 2022, 12, 4916. [Google Scholar] [CrossRef]
- Lederer, C.; Mayer, K.; Somogyi, V.; Kriegsmann, K.; Kriegsmann, M.; Buschulte, K.; Polke, M.; Findeisen, P.; Herth, F.; Kreuter, M. Krebs von Den Lungen-6 as a Potential Predictive Biomarker in Fibrosing Interstitial Lung Diseases. Respiration 2023, 102, 591–600. [Google Scholar] [CrossRef]
- Huang, T.-H.; Kuo, C.-W.; Chen, C.-W.; Tseng, Y.-L.; Wu, C.-L.; Lin, S.-H. Baseline Plasma KL-6 Level Predicts Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Nintedanib: A Retrospective Real-World Cohort Study. BMC Pulm. Med. 2021, 21, 165. [Google Scholar] [CrossRef]
- Nakano, A.; Ohkubo, H.; Fukumitsu, K.; Fukuda, S.; Kanemitsu, Y.; Takemura, M.; Maeno, K.; Ito, Y.; Oguri, T.; Niimi, A. Remarkable Improvement in a Patient with Idiopathic Pulmonary Fibrosis after Treatment with Nintedanib. Intern. Med. 2019, 58, 1141–1144. [Google Scholar] [CrossRef]
- Landi, C.; Bergantini, L.; Cameli, P.; d’Alessandro, M.; Carleo, A.; Shaba, E.; Rottoli, P.; Bini, L.; Bargagli, E. Idiopathic Pulmonary Fibrosis Serum Proteomic Analysis before and after Nintedanib Therapy. Sci. Rep. 2020, 10, 9378. [Google Scholar] [CrossRef]
- Devendra, G.; Spragg, R.G. Lung Surfactant in Subacute Pulmonary Disease. Respir. Res. 2002, 3, 11–19. [Google Scholar] [CrossRef]
- Carreto-Binaghi, L.E.; Aliouat, E.M.; Taylor, M.L. Surfactant Proteins, SP-A and SP-D, in Respiratory Fungal Infections: Their Role in the Inflammatory Response. Respir. Res. 2016, 17, 66. [Google Scholar] [CrossRef]
- Kishore, U.; Greenhough, T.J.; Waters, P.; Shrive, A.K.; Ghai, R.; Kamran, M.F.; Bernal, A.L.; Reid, K.B.M.; Madan, T.; Chakraborty, T. Surfactant Proteins SP-A and SP-D: Structure, Function and Receptors. Mol. Immunol. 2006, 43, 1293–1315. [Google Scholar] [CrossRef] [PubMed]
- Pastva, A.M.; Wright, J.R.; Williams, K.L. Immunomodulatory Roles of Surfactant Proteins A and D: Implications in Lung Disease. Proc. Am. Thorac. Soc. 2007, 4, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Korfei, M.; Mahavadi, P.; Beck D von der Ruppert, C.; Markart, P. Unravelingthe progressive pathophysiology of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2012, 21, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A. The Molecular Era of Surfactant Biology. Neonatology 2014, 105, 337–343. [Google Scholar] [CrossRef]
- Sorensen, G.L.; Husby, S.; Holmskov, U. Surfactant protein A and surfactant protein D variation in pulmonary disease. Immunobiology 2007, 212, 381–416. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Otsuka, M.; Chiba, H.; Ikeda, K.; Mori, Y.; Umeda, Y.; Nishikiori, H.; Kuronuma, K.; Takahashi, H. Surfactant Protein A as a Biomarker of Outcomes of Anti-Fibrotic Drug Therapy in Patients with Idiopathic Pulmonary Fibrosis. BMC Pulm. Med. 2020, 20, 27. [Google Scholar] [CrossRef]
- Ikeda, K.; Pirfenidone Clinical Study Group in Japan; Chiba, H.; Nishikiori, H.; Azuma, A.; Kondoh, Y.; Ogura, T.; Taguchi, Y.; Ebina, M.; Sakaguchi, H.; et al. Serum Surfactant Protein D as a Predictive Biomarker for the Efficacy of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis: A Post-Hoc Analysis of the Phase 3 Trial in Japan. Respir. Res. 2020, 21, 316. [Google Scholar] [CrossRef]
- Hill, D.B.; Button, B.; Rubinstein, M.; Boucher, R.C. Physiology and Pathophysiology of Human Airway Mucus. Physiol. Rev. 2022, 102, 1757–1836. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Qu, D.; Yu, J.; Yang, J. The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis ConsideringMUC5B. BioMed Res. Int. 2019, 2019, 9712464. [Google Scholar] [CrossRef]
- Yang, I.V.; Fingerlin, T.E.; Evans, C.M.; Schwarz, M.I.; Schwartz, D.A. MUC5B and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S2), S193–S199. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Yang, I.V.; Walts, A.D.; Watson, A.M.; Helling, B.A.; Fletcher, A.A.; Lara, A.R.; Schwarz, M.I.; Evans, C.M.; Schwartz, D.A. MUC5B Promoter Variant Rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Biondini, D.; Cocconcelli, E.; Bernardinello, N.; Lorenzoni, G.; Rigobello, C.; Lococo, S.; Castelli, G.; Baraldo, S.; Cosio, M.G.; Gregori, D.; et al. Prognostic Role of MUC5B Rs35705950 Genotype in Patients with Idiopathic Pulmonary Fibrosis (IPF) on Antifibrotic Treatment. Respir. Res. 2021, 22, 98. [Google Scholar] [CrossRef] [PubMed]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Grove Villalon, D.E.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef]
- Doubkova, M.; Kriegova, E.; Littnerova, S.; Schneiderova, P.; Sterclova, M.; Bartos, V.; Plackova, M.; Zurkova, M.; Bittenglova, R.; Lostaková, V.; et al. DSP Rs2076295 Variants Influence Nintedanib and Pirfenidone Outcomes in Idiopathic Pulmonary Fibrosis: A Pilot Study. Ther. Adv. Respir. Dis. 2021, 15, 175346662110425. [Google Scholar] [CrossRef]
- Jo, H.E.; Troy, L.K.; Keir, G.; Chambers, D.C.; Holland, A.; Goh, N.; Wilsher, M.; de Boer, S.; Moodley, Y.; Grainge, C.; et al. Treatment of Idiopathic Pulmonary Fibrosis in Australia and New Zealand: A Position Statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia: Treatment of IPF. Respirology 2017, 22, 1436–1458. [Google Scholar] [CrossRef]
- Xu, Y.; Zhong, W.; Zhang, L.; Zhao, J.; Li, L.; Wang, M. Clinical Characteristics of Patients with Lung Cancer and Idiopathic Pulmonary Fibrosis in China: Lung Cancer and Idiopathic Pulmonary Fibrosis. Thorac. Cancer 2012, 3, 156–161. [Google Scholar] [CrossRef]
- Kwon, B.S.; Kim, E.S.; Lim, S.Y.; Song, M.J.; Kim, Y.W.; Kim, H.-J.; Lee, Y.J.; Park, J.S.; Cho, Y.-J.; Yoon, H.I.; et al. The Significance of Elevated Tumor Markers among Patients with Interstitial Lung Diseases. Sci. Rep. 2022, 12, 16702. [Google Scholar] [CrossRef]
- Ricci, A.; Mariotta, S.; Bronzetti, E.; Bruno, P.; Vismara, L.; De Dominicis, C.; Laganà, B.; Paone, G.; Mura, M.; Rogliani, P.; et al. Serum CA 15-3 Is Increased in Pulmonary Fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 54–63. [Google Scholar]
- Moll, S.A.; Wiertz, I.A.; Vorselaars, A.D.M.; Zanen, P.; Ruven, H.J.T.; van Moorsel, C.H.M.; Grutters, J.C. Serum Biomarker CA 15-3 as Predictor of Response to Antifibrotic Treatment and Survival in Idiopathic Pulmonary Fibrosis. Biomark. Med. 2020, 14, 997–1007. [Google Scholar] [CrossRef]
- Kruit, A.; Gerritsen, W.B.M.; Pot, N.; Grutters, J.C.; van den Bosch, J.M.M.; Ruven, H.J.T. CA 15-3 as an Alternative Marker for KL-6 in Fibrotic Lung Diseases. Sarcoidosis Vasc. Diffus. Lung Dis. 2010, 27, 138–146. [Google Scholar]
- Ji, X.Y.; Jin, P.; Yu, G.; Wang, Q.-Q.; Yu, P. Serum biomarkers in evaluation and management of idiopathic pulmonary fibrosis. Precis. Med. Res. 2020, 2, 127–135. [Google Scholar] [CrossRef]
- Rusanov, V.; Kramer, M.R.; Raviv, Y.; Medalion, B.; Guber, A.; Shitrit, D. The Significance of Elevated Tumor Markers among Patients with Idiopathic Pulmonary Fibrosis before and after Lung Transplantation. Chest 2012, 141, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujimoto, D.; Uehara, K.; Shimizu, R.; Ito, J.; Kogo, M.; Teraoka, S.; Kato, R.; Nagata, K.; Nakagawa, A.; et al. The Prognostic Value of Serum CA 19-9 for Patients with Advanced Lung Adenocarcinoma. BMC Cancer 2016, 16, 890. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, D.-K.; Lee, M.-Y.; Lim, H.-S.; Kwon, M.-J.; Lee, Y.-T.; Yoon, K.J.; Park, C.-H. The Association of Carbohydrate Antigen (CA) 19-9 Levels and Low Skeletal Muscle Mass in Healthy Adults. Nutrients 2023, 15, 3394. [Google Scholar] [CrossRef]
- Lee, T.; Teng, T.Z.J.; Shelat, V.G. Carbohydrate Antigen 19-9—Tumor Marker: Past, Present, and Future. World J. Gastrointest. Surg. 2020, 12, 468–490. [Google Scholar] [CrossRef]
- Balestro, E.; Castelli, G.; Bernardinello, N.; Cocconcelli, E.; Biondini, D.; Fracasso, F.; Rea, F.; Saetta, M.; Baraldo, S.; Spagnolo, P. CA 19-9 Serum Levels in Patients with End-Stage Idiopathic Pulmonary Fibrosis (IPF) and Other Interstitial Lung Diseases (ILDs): Correlation with Functional Decline. Chron. Respir. Dis. 2020, 17, 147997312095842. [Google Scholar] [CrossRef]
- Majewski, S.; Szewczyk, K.; Żal, A.; Białas, A.J.; Miłkowska-Dymanowska, J.; Piotrowski, W.J. Serial Measurements of Circulating KL-6, SP-D, MMP-7, CA19-9, CA-125, CCL18, and Periostin in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Therapy: An Exploratory Study. J. Clin. Med. 2021, 10, 3864. [Google Scholar] [CrossRef]
- Ay, S.; Ozyukseler, D.T.; Dulgar, O.; Basak, M.; Yildirim, M.E.; Gumus, M. Initial ca 125 Value as a Predictive Marker for High-Grade Serous Ovarian Cancer. J. Coll. Physicians Surg. Pak. 2021, 30, 651–656. [Google Scholar] [CrossRef]
- Kumric, M.; Kurir, T.T.; Bozic, J.; Glavas, D.; Saric, T.; Marcelius, B.; D’Amario, D.; Borovac, J.A. Carbohydrate Antigen 125: A Biomarker at the Crossroads of Congestion and Inflammation in Heart Failure. Card. Fail. Rev. 2021, 7, e19. [Google Scholar] [CrossRef]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; Fahy, W.A.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An Epithelial Biomarker Signature for Idiopathic Pulmonary Fibrosis: An Analysis from the Multicentre PROFILE Cohort Study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Alqalyoobi, S.; Linderholm, A.; Bowman, W.S.; Lee, C.T.; Pugashetti, J.V.; Sarma, N.; Ma, S.-F.; Haczku, A.; Sperling, A.; et al. Circulating Plasma Biomarkers of Survival in Antifibrotic-Treated Patients with Idiopathic Pulmonary Fibrosis. Chest 2020, 158, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Clarke, L.; Pal, A.; Buchwald, P.; Eglinton, T.; Wakeman, C.; Frizelle, F. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Ann. Coloproctol. 2019, 35, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Rule, A.H.; Goleski-Reilly, C.; Sachar, D.B.; Vandevoorde, J.; Janowitz, H.D. Circulating Carcinoembryonic Antigen (CEA): Relationship to Clinical Status of Patients with Inflammatory Bowel Disease. Gut 1973, 14, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.S.; Lee, J.G.; Paik, H.C.; Kim, S.J.; Lee, S.; Kim, S.Y.; Park, M.S.; Haam, S. Carcinoembryonic Antigen Predicts Waitlist Mortality in Lung Transplant Candidates with Idiopathic Pulmonary Fibrosis. Eur. J. Cardiothorac. Surg. 2018, 54, 847–852. [Google Scholar] [CrossRef]
- Fahim, A.; Crooks, M.G.; Wilmot, R.; Campbell, A.P.; Morice, A.H.; Hart, S.P. Serum Carcinoembryonic Antigen Correlates with Severity of Idiopathic Pulmonary Fibrosis: Serum CEA and IPF. Respirology 2012, 17, 1247–1252. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, M.; Huang, H.; Jiang, X.; Gong, K.; Liu, Y.; Kuang, X.; Yang, X. Serum Carcinoembryonic Antigen Elevation in Benign Lung Diseases. Sci. Rep. 2021, 11, 19044. [Google Scholar] [CrossRef]
- Winkelmann, A.; Noack, T. The Clara Cell: A “Third Reich Eponym”? Eur. Respir. J. 2010, 36, 722–727. [Google Scholar] [CrossRef]
- Singh, G.; Katyal, S.L. Clara Cells and Clara Cell 10 KD Protein (CC10). Am. J. Respir. Cell Mol. Biol. 1997, 17, 141–143. [Google Scholar] [CrossRef]
- Bernard, A.; Hermans, C.; Van Houte, G. Transient Increase of Serum Clara Cell Protein (CC16) after Exposure to Smoke. Occup. Environ. Med. 1997, 54, 63–65. [Google Scholar] [CrossRef]
- Rokicki, W.; Rokicki, M.; Wojtacha, J.; Dżeljijli, A. The Role and Importance of Club Cells (Clara Cells) in the Pathogenesis of Some Respiratory Diseases. Kardiochir. Torakochirurgia Pol. 2016, 1, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.P.; Keating, A.; Waddell, T.K. Airway Regeneration: The Role of the Clara Cell Secretory Protein and the Cells That Express It. Cytotherapy 2009, 11, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Almuntashiri, S.; Zhu, Y.; Han, Y.; Wang, X.; Somanath, P.R.; Zhang, D. Club Cell Secreted Protein CC16: Potential Applications in Prognosis and Therapy for Pulmonary Diseases. J. Clin. Med. 2020, 9, 4039. [Google Scholar] [CrossRef] [PubMed]
- Hermans, C.; Aly, O.; Nyberg, B.-I.; Peterson, C.; Bernard, A. Determinants of Clara Cell Protein (CC16) Concentration in Serum: A Reassessment with Two Different Immunoassays. Clin. Chim. Acta 1998, 272, 101–110. [Google Scholar] [CrossRef]
- Buendía-Roldán, I.; Ruiz, V.; Sierra, P.; Montes, E.; Ramírez, R.; Vega, A.; Salgado, A.; Vargas, M.H.; Mejía, M.; Pardo, A.; et al. Increased Expression of CC16 in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0168552. [Google Scholar] [CrossRef] [PubMed]
- Gampawar, P.; Schmidt, R.; Schmidt, H. Telomere Length and Brain Aging: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2022, 80, 101679. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.; Boonekamp, J. Does Oxidative Stress Shorten Telomeres in Vivo? A Meta-Analysis. Ageing Res. Rev. 2023, 85, 101854. [Google Scholar] [CrossRef]
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef]
- Kordinas, V.; Ioannidis, A.; Chatzipanagiotou, S. The Telomere/Telomerase System in Chronic Inflammatory Diseases. Cause or Effect? Genes 2016, 7, 60. [Google Scholar] [CrossRef]
- Borges, G.; Criqui, M.; Harrington, L. Tieing Together Loose Ends: Telomere Instability in Cancer and Aging. Mol. Oncol. 2022, 16, 3380–3396. [Google Scholar] [CrossRef]
- Verma, A.K.; Singh, P.; Al-Saeed, F.A.; Ahmed, A.E.; Kumar, S.; Kumar, A.; Dev, K.; Dohare, R. Unravelling the Role of Telomere Shortening with Ageing and Their Potential Association with Diabetes, Cancer, and Related Lifestyle Factors. Tissue Cell 2022, 79, 101925. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Planas-Cerezales, L.; Perona, R. Telomere Shortening in Idiopathic Pulmonary Fibrosis. Arch. Bronconeumol. 2018, 54, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Cai, H.; Li, H.; Zhuang, Y.; Min, H.; Wen, Y.; Yang, J.; Gao, Q.; Shi, Y.; Yi, L. Association between Telomere Length and Survival in Patients with Idiopathic Pulmonary Fibrosis. Respirology 2015, 20, 947–952. [Google Scholar] [CrossRef]
- Bilgili, H.; Białas, A.J.; Górski, P.; Piotrowski, W.J. Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2019, 8, 1232. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-I.; Ryerson, C.J.; Lee, J.S.; Kukreja, J.; Barry, S.S.; Jones, K.D.; Elicker, B.M.; Kim, D.S.; Papa, F.R.; Collard, H.R.; et al. Cleaved Cytokeratin-18 Is a Mechanistically Informative Biomarker in Idiopathic Pulmonary Fibrosis. Respir. Res. 2012, 13, 105. [Google Scholar] [CrossRef]
- Decaris, M.L.; Schaub, J.R.; Chen, C.; Cha, J.; Lee, G.G.; Rexhepaj, M.; Ho, S.S.; Rao, V.; Marlow, M.M.; Kotak, P.; et al. Dual inhibition of αvβ6 and αvβ1 reduces fibrogenesis in lung tissue explants from patients with IPF. Respir. Res. 2021, 22, 265. [Google Scholar] [CrossRef]
- Koivisto, L.; Bi, J.; Häkkinen, L.; Larjava, H. Integrin Avβ6: Structure, Function and Role in Health and Disease. Int. J. Biochem. Cell Biol. 2018, 99, 186–196. [Google Scholar] [CrossRef]
- John, A.E.; Luckett, J.C.; Tatler, A.L.; Awais, R.O.; Desai, A.; Habgood, A.; Ludbrook, S.; Blanchard, A.D.; Perkins, A.C.; Jenkins, R.G.; et al. Preclinical SPECT/CT Imaging of Avβ6 Integrins for Molecular Stratification of Idiopathic Pulmonary Fibrosis. J. Nucl. Med. 2013, 54, 2146–2152. [Google Scholar] [CrossRef]
- John, A.E.; Graves, R.H.; Pun, K.T.; Vitulli, G.; Forty, E.J.; Mercer, P.F.; Morrell, J.L.; Barrett, J.W.; Rogers, R.F.; Hafeji, M.; et al. Translational Pharmacology of an Inhaled Small Molecule Avβ6 Integrin Inhibitor for Idiopathic Pulmonary Fibrosis. Nat. Commun. 2020, 11, 4659. [Google Scholar] [CrossRef]
- Maher, T.M.; Simpson, J.K.; Porter, J.C.; Wilson, F.J.; Chan, R.; Eames, R.; Cui, Y.; Siederer, S.; Parry, S.; Kenny, J.; et al. A Positron Emission Tomography Imaging Study to Confirm Target Engagement in the Lungs of Patients with Idiopathic Pulmonary Fibrosis Following a Single Dose of a Novel Inhaled Avβ6 Integrin Inhibitor. Respir. Res. 2020, 21, 75. [Google Scholar] [CrossRef]
- Saini, G.; Porte, J.; Weinreb, P.H.; Violette, S.M.; Wallace, W.A.; McKeever, T.M.; Jenkins, G. Avβ6 Integrin May Be a Potential Prognostic Biomarker in Interstitial Lung Disease. Eur. Respir. J. 2015, 46, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.L.; Murray, L.A.; Crestani, B.; Sleeman, M.A. Is Personalised Medicine the Key to Heterogeneity in Idiopathic Pulmonary Fibrosis? Pharmacol. Ther. 2017, 169, 35–46. [Google Scholar] [CrossRef]
- Kulkarni, T.; de Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar Epithelial Disintegrity in Pulmonary Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Xiao, H.; Dong, R.; Geng, J.; Xie, B.; Ren, Y.; Dai, H. Krebs von Den Lungen-6 Levels in Untreated Idiopathic Pulmonary Fibrosis. Clin. Respir. J. 2022, 16, 234–243. [Google Scholar] [CrossRef]
- Zheng, P.; Zheng, X.; Takehiro, H.; Cheng, Z.J.; Wang, J.; Xue, M.; Lin, Q.; Huang, Z.; Huang, H.; Liao, C.; et al. The Prognostic Value of Krebs von Den Lungen-6 and Surfactant Protein-A Levels in the Patients with Interstitial Lung Disease. J. Transl. Int. Med. 2021, 9, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Kim, J.; Cho, H.S.; Kim, H.C. Baseline Serum Krebs von Den Lungen-6 as a Biomarker for the Disease Progression in Idiopathic Pulmonary Fibrosis. Sci. Rep. 2022, 12, 8564. [Google Scholar] [CrossRef]
- Kuroki, Y.; Takahashi, H.; Chiba, H.; Akino, T. Surfactant Proteins A and D: Disease Markers. Biochim. Biophys. Acta Mol. Basis Dis. 1998, 1408, 334–345. [Google Scholar] [CrossRef]
- Takahashi, H.; Fujishima, T.; Koba, H.; Murakami, S.; Kurokawa, K.; Shibuya, Y.; Shiratori, M.; Kuroki, Y.; Abe, S. Serum Surfactant Proteins A and D as Prognostic Factors in Idiopathic Pulmonary Fibrosis and Their Relationship to Disease Extent. Am. J. Respir. Crit. Care Med. 2000, 162, 1109–1114. [Google Scholar] [CrossRef]
- Ley, B.; Brown, K.K.; Collard, H.R. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L681–L691. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, L.; Cong, Y.-S. Telomere Dysfunction in Idiopathic Pulmonary Fibrosis. Front. Med. 2021, 8, 739810. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamfir, A.-S.; Zabara, M.L.; Arcana, R.I.; Cernomaz, T.A.; Zabara-Antal, A.; Marcu, M.T.D.; Trofor, A.; Zamfir, C.L.; Crișan-Dabija, R. Exploring the Role of Biomarkers Associated with Alveolar Damage and Dysfunction in Idiopathic Pulmonary Fibrosis—A Systematic Review. J. Pers. Med. 2023, 13, 1607. https://doi.org/10.3390/jpm13111607
Zamfir A-S, Zabara ML, Arcana RI, Cernomaz TA, Zabara-Antal A, Marcu MTD, Trofor A, Zamfir CL, Crișan-Dabija R. Exploring the Role of Biomarkers Associated with Alveolar Damage and Dysfunction in Idiopathic Pulmonary Fibrosis—A Systematic Review. Journal of Personalized Medicine. 2023; 13(11):1607. https://doi.org/10.3390/jpm13111607
Chicago/Turabian StyleZamfir, Alexandra-Simona, Mihai Lucian Zabara, Raluca Ioana Arcana, Tudor Andrei Cernomaz, Andreea Zabara-Antal, Marius Traian Dragoș Marcu, Antigona Trofor, Carmen Lăcrămioara Zamfir, and Radu Crișan-Dabija. 2023. "Exploring the Role of Biomarkers Associated with Alveolar Damage and Dysfunction in Idiopathic Pulmonary Fibrosis—A Systematic Review" Journal of Personalized Medicine 13, no. 11: 1607. https://doi.org/10.3390/jpm13111607
APA StyleZamfir, A. -S., Zabara, M. L., Arcana, R. I., Cernomaz, T. A., Zabara-Antal, A., Marcu, M. T. D., Trofor, A., Zamfir, C. L., & Crișan-Dabija, R. (2023). Exploring the Role of Biomarkers Associated with Alveolar Damage and Dysfunction in Idiopathic Pulmonary Fibrosis—A Systematic Review. Journal of Personalized Medicine, 13(11), 1607. https://doi.org/10.3390/jpm13111607