

Challenges and Opportunities in the Genetic Analysis of Inherited Retinal Dystrophies in Africa, a Literature Review




Abstract
:1. Introduction
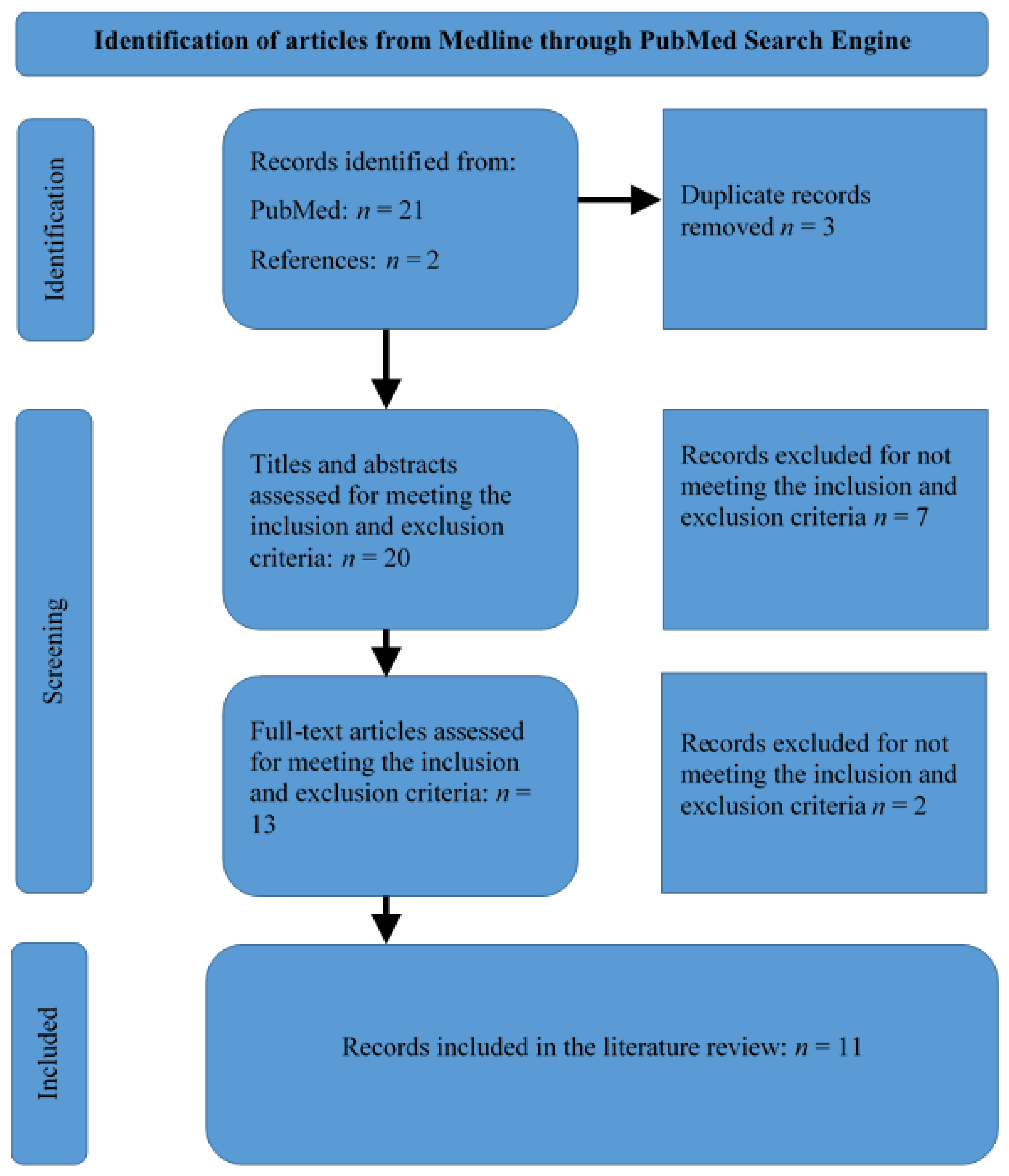
2. Materials and Methods
3. Results
3.1. Summary of Selected Articles
3.2. Testing Methods Used
3.3. Common Genetically Diagnosed Diseases
3.3.1. Retinitis Pigmentosa
3.3.2. Leber Congenital Amaurosis (LCA)
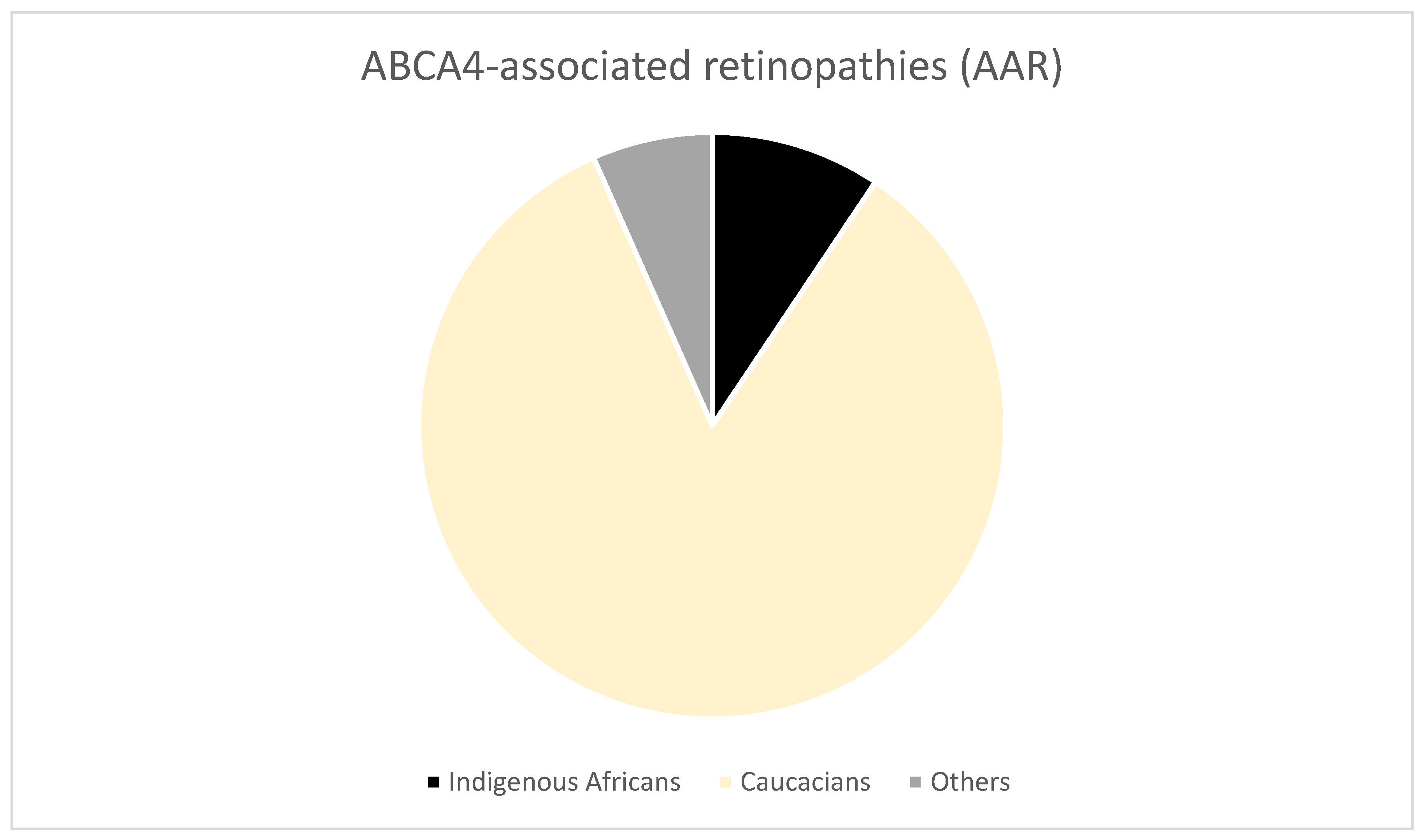
3.3.3. Stargardt Disease (STGD)
3.3.4. Cone Dystrophy
3.3.5. Syndromic IRDs
3.4. Diagnostic Challenges
3.5. Diagnostic Opportunities
4. Discussion
4.1. Genes Affected in the Most Prevalent IRDs
4.2. Choice of Test
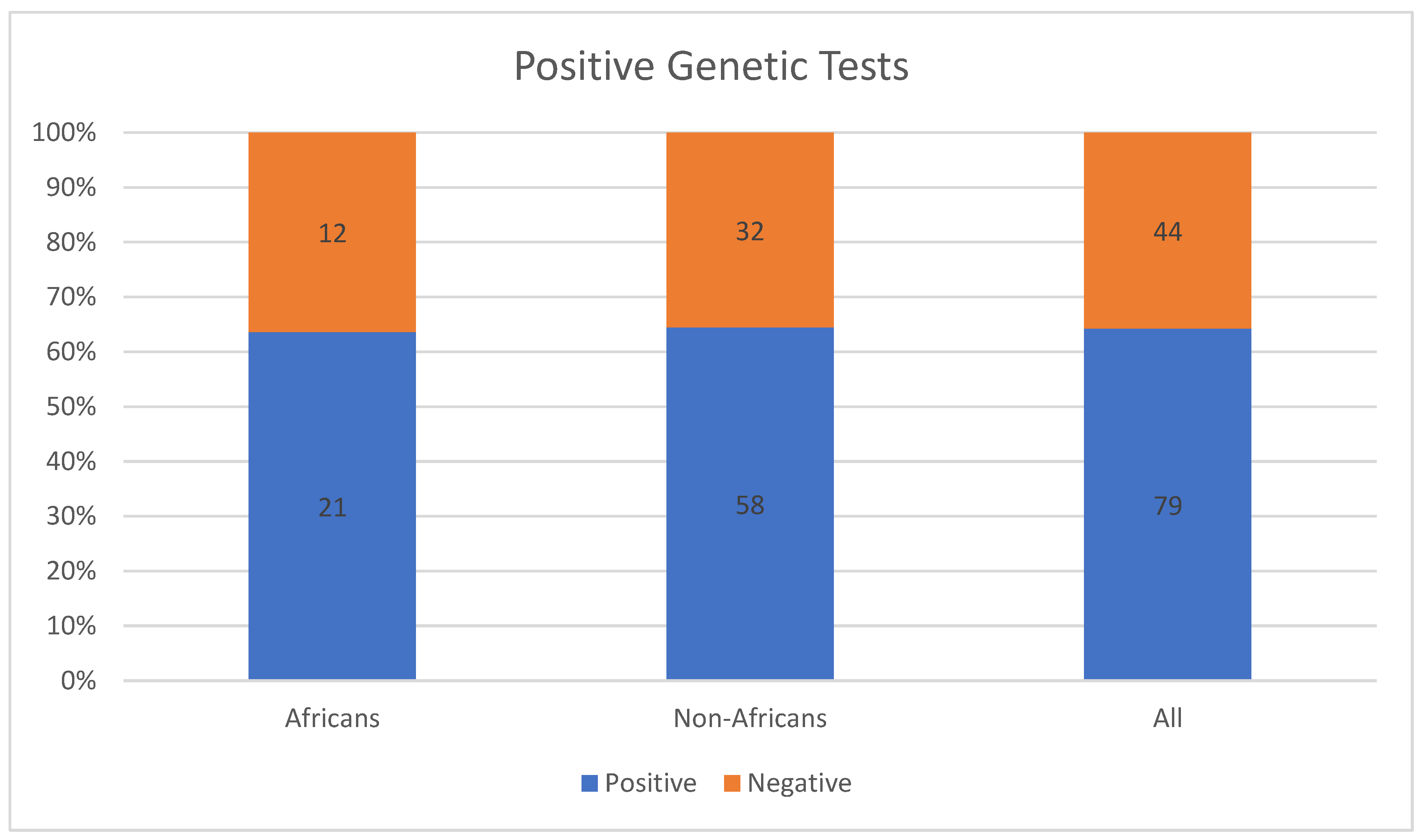
4.3. Uniqueness of IRD-Causing Mutations among Africans
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henderson, R.H. Inherited retinal dystrophies. Paediatr. Child Health 2020, 30, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Moshiri, A.; Chen, R.; Kim, S.; Harris, R.A.; Li, Y.; Raveendran, M.; Davis, S.; Liang, Q.; Pomerantz, O.; Wang, J.; et al. A nonhuman primate model of inherited retinal disease. J. Clin. Investig. 2019, 129, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.; Rebello, G.; Greenberg, J.; Ramesar, R. Update on Inherited Retinal Disease in South Africa: Encouraging Diversity in Molecular Genetics. Adv. Exp. Med. Biol. 2019, 1185, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Goliath, R.; Rebello, G.; Bardien, S.; September, A.V.; Bartmann, L.; Loubser, F.; Greenberg, L.J.; Ramesar, R.S. Inherited retinal disorders in South Africa and the clinical impact of evolving technologies. S. Afr. Med. J. 2016, 106, S33–S37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the epidemiology of age-related macular degeneration. Asia-Pacific J. Ophthalmol. 2017, 6, 493–497. [Google Scholar] [CrossRef]
- Aouadj, C.; Banhazi, J.; Shaikh, J.; Thakker, D.; Lacey, S.; Viriato, D.; Fischer, M. PSY28-Epidemiology of RPE65 gene mutation related inherited retinal dystrophies: A systematic literature review. Value Health 2018, 21, S440. [Google Scholar] [CrossRef] [Green Version]
- Sallum, J.M.F.; Kaur, V.P.; Shaikh, J.; Banhazi, J.; Spera, C.; Aouadj, C.; Viriato, D.; Fischer, M.D. Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review. Adv. Ther. 2022, 39, 1179–1198. [Google Scholar] [CrossRef]
- Roberts, L.; Ratnapriya, R.; Du Plessis, M.; Chaitankar, V.; Ramesar, R.S.; Swaroop, A. Molecular diagnosis of inherited retinal diseases in indigenous African populations by whole-exome sequencing. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6374–6381. [Google Scholar] [CrossRef] [Green Version]
- Ellingford, J.; Barton, S.; Bhaskar, S.; Williams, S.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.; Perveen, R.; Hall, G.; Newman, W.; et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Cornish, E.E.; Vaze, A.; Jamieson, R.V.; Grigg, J.R. The electroretinogram in the genomics era: Outer retinal disorders. Eye 2021, 35, 2406–2418. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Gene discovery and prevalence in inherited retinal dystrophies. Comptes Rendus Biol. 2014, 337, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Leroy, B.P.; Black, G.; Ong, T.; Yoon, D.; Trzupek, K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J. Rare Dis. 2021, 16, 514. [Google Scholar] [CrossRef]
- Bouzidi, A.; Charoute, H.; Charif, M.; Amalou, G.; Kandil, M.; Barakat, A.; Lenaers, G. Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies. Orphanet J. Rare Dis. 2022, 17, 197. [Google Scholar] [CrossRef]
- Maltese, P.E.; Colombo, L.; Martella, S.; Rossetti, L.; El Shamieh, S.; Sinibaldi, L.; Passarelli, C.; Coppè, A.M.; Buzzonetti, L.; Falsini, B.; et al. Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals. Front. Genet. 2022, 13, 914345. [Google Scholar] [CrossRef]
- Bouzidi, A.; Charif, M.; Bouzidi, A.; Amalou, G.; Kandil, M.; Barakat, A.; Lenaers, G. Clinical and genetic investigations of three moroccan families with retinitis pigmentosa phenotypes. Mol. Vis. 2021, 27, 17–25. Available online: http://www.molvis.org/molvis/v27/17 (accessed on 24 December 2022). [PubMed]
- Greenberg, J.; Bartmann, L.; Ramesar, R.; Beighton, P. Retinitis pigmentosa in Southern Africa. Clin. Genet. 1993, 44, 232–235. [Google Scholar] [CrossRef]
- Perrault, I.; Rozet, J.M.; Calvas, P.; Gerber, S.; Camuzat, A.; Dollfus, H.; Châtelin, S.; Souied, E.; Ghazi, I.; Leowski, C.; et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 1996, 14, 461–464. [Google Scholar] [CrossRef]
- Maggi, J.; Roberts, L.; Koller, S.; Rebello, G.; Berger, W.; Ramesar, R. De novo assembly-based analysis of RPGR exon ORF15 in an indigenous African cohort overcomes limitations of a standard next-generation sequencing (NGS) data analysis pipeline. Genes 2020, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.B.; McHale, J.C.; Keen, T.J.; Tarttelin, E.E.; Goliath, R.; Van Lith-Verhoeven, J.J.; Greenberg, J.; Ramesar, R.S.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet. 2001, 10, 1555–1562. [Google Scholar] [CrossRef]
- Camuzat, A.; Rozet, J.-M.; Dollfus, H.; Gerber, S.; Perrault, I.; Weissenbach, J.; Munnich, A.; Kaplan, J. Evidence for genetic heterogeneity in Leber’s congenital amaurosis and fine mapping of LCA1 to chromosome 17P13. Human. Genet. 1996, 97, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Falfoul, Y.; Turki, A.; Habibi, I.; Hassairi, A. Clinical and genetic characteristics of enhanced S-cone syndrome in a Tunisian cohort: Experience of the oculogenetic laboratory LR14SP01. Acta Ophthalmol. 2018, 96, F116. [Google Scholar]
- Roberts, L.J.; Nossek, C.A.; Greenberg, L.J.; Ramesar, R.S. Stargardt macular dystrophy: Common ABCA4 mutations in South Africa-establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 2012, 18, 280–289. Available online: http://www.molvis.org/molvis/v18/a32 (accessed on 24 December 2022). [PubMed]
- September, A.V.; Vorster, A.A.; Ramesar, R.S.; Greenberg, L.J. Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Gurdasani, D.; Carstensen, T.; Tekola-Ayele, F.; Pagani, L.; Tachmazidou, I.; Hatzikotoulas, K.; Karthikeyan, S.; Iles, L.; Pollard, M.O.; Choudhury, A.; et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2015, 517, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Jespersgaard, C.; Bertelsen, M.; Arif, F.; Gellert-Kristensen, H.G.; Fang, M.; Jensen, H.; Rosenberg, T.; Tümer, Z.; Møller, L.B.; Brøndum-Nielsen, K.; et al. Bi-allelic pathogenic variations in mertk including deletions are associated with an early onset progressive form of retinitis pigmentosa. Genes 2020, 11, 1517. [Google Scholar] [CrossRef]
- Poli, F.E.; Yusuf, I.H.; Clouston, P.; Shanks, M.; Whitfield, J.; Issa, P.C.; MacLaren, R.E. MERTK missense variants in three patients with retinitis pigmentosa. Ophthalmic Genet. 2022. [Google Scholar] [CrossRef]
- Kuehlewein, L.; Zobor, D.; Stingl, K.; Kempf, M.; Nasser, F.; Bernd, A.; Biskup, S.; Cremers, F.; Khan, M.; Mazzola, P.; et al. Clinical phenotype of pde6b-associated retinitis pigmentosa. Int. J. Mol. Sci. 2021, 22, 2374. [Google Scholar] [CrossRef]
- Downes, S.M.; Nguyen, T.; Tai, V.; Broadgate, S.; Shah, M.; Al-Khuzaei, S.; MacLaren, R.E.; Shanks, M.; Clouston, P.; Halford, S. Genetic and clinical findings in an ethnically diverse cohort with retinitis pigmentosa associated with pathogenic variants in cerkl. Genes 2020, 11, 1497. [Google Scholar] [CrossRef]
- Cremers, F.P.; Lee, W.; Collin, R.W.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Yang, C.-M.; Yang, C.-H.; Hou, Y.-C.; Chen, T.-C. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes 2021, 12, 1261. [Google Scholar] [CrossRef]
- Kondkar, A.A.; Abu-Amero, K.K. Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. Exp. Eye Res. 2019, 189, 107834. [Google Scholar] [CrossRef] [PubMed]
- Doucette, L.P.; Noel, N.C.L.; Zhai, Y.; Xu, M.; Caluseriu, O.; Hoang, S.C.; Radziwon, A.J.; MacDonald, I.M. Whole exome sequencing reveals putatively novel associations in retinopathies and drusen formation. Eur. J. Hum. Genet. 2021, 29, 1171–1185. [Google Scholar] [CrossRef]
- Khan, M.; Arno, G.; Fakin, A.; Parfitt, D.A.; Dhooge, P.P.; Albert, S.; Bax, N.M.; Duijkers, L.; Niblock, M.; Hau, K.L.; et al. Detailed Phenotyping and Therapeutic Strategies for Intronic ABCA4 Variants in Stargardt Disease. Mol. Ther. Nucleic Acids 2020, 21, 412–427. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002733. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, T.A.C.D.; Georgiou, M.; Robson, A.G.; Michaelides, M. KCNV2 retinopathy: Clinical features, molecular genetics and directions for future therapy. Ophthalmic Genet. 2020, 41, 208–215. [Google Scholar] [CrossRef]
- Yaylacıoğlu, F. Genetic testing strategies in inherited retinal diseases: Practical aspects for ophthalmologists. J. Retin. 2022, 31, 193. [Google Scholar] [CrossRef]
- Ganapathi, M.; Thomas-Wilson, A.; Buchovecky, C.; Dharmadhikari, A.; Barua, S.; Lee, W.; Ruan, M.Z.C.; Soucy, M.; Ragi, S.; Tanaka, J.; et al. Clinical exome sequencing for inherited retinal degenerations at a tertiary care center. Sci. Rep. 2022, 12, 9358. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.S.; Procopio, R.; Davila, H.J.; Bello, N.; Ku, C.; Pennesi, M.E.; Yang, P.; Nagiel, A.; Mahroo, O.A.; Aleman, T.S.; et al. Inherited Retinal Disease Panels—Caveat Emptor—Truly Know Your Inherited Retinal Disease Panel. Retina 2022, 42, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zampaglione, E.; Maher, M.; Place, E.M.; Wagner, N.E.; DiTroia, S.; Chao, K.R.; England, E.; Cmg, B.; Catomeris, A.; Nassiri, S.; et al. The Importance of Automation in Genetic Diagnosis: Lessons from Analyzing an Inherited Retinal Degeneration Cohort with the Mendelian Analysis Toolkit (MATK). medRxiv 2022, 24, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Division of Human Genetics. Retinal Degeneration. Available online: http://www.humangenetics.uct.ac.za/hg/research/retinal_degeneration (accessed on 24 December 2022).




{kind=link}
{kind=link}
{kind=link}
| Authors | Location of patients | Diseases | Included Africans | Genetic Testing Methods Used |
|---|---|---|---|---|
| Bouzidi et al. [14] | North Africa | IRDs and inherited optical neuropathies (ION) | 413 families | Next-generation sequencing |
| Maltese et al. [15] | North Africa | IRDs | 33 patients | Next-generation sequencing (NGS) and gene sequencing panels targeting a specific set of genes, Sanger sequencing and—when necessary—multiplex ligation-dependent probe amplification (MLPA) |
| Bouzidi et al. [16] | Morocco | Retinitis pigmentosa | 3 families | Whole exome sequencing |
| Greenberg et al. [17] | South Africa | Retinitis pigmentosa | 75 families | Unspecified DNA banking methods |
| Perrault et al. [18] | North Africa | Leber congenital amaurosis | 7 families | Sequencing using primers specific to the cDNA sequence and mutation screening |
| Maggi et al. [19] | South Africa | Retinitis pigmentosa and recessive macular degeneration | RP: 17 patients MD: 15 probands | Next-generation PCR and sequencing |
| McKie et al. [20] | South Africa | Retinitis pigmentosa | 1 family | PCR, segregation analysis, reverse transcriptase PCR, and Nothern blot analysis |
| Camuzat et al. [21] | North Africa | Leber congenital amaurosis | 7 families | Genotyping using PCR applying hypervariable microsatellites |
| Falfoul et al. [22] | Tunisia | Enhance-S-cone syndrome | 6 families | Unspecified genetic tests |
| Roberts et al. [23] | South Africa | ABCA4-associated retinopathies | 17 probands | Rapid test to detect seven ABCA4 mutations |
| September et al. [24] | South Africa | Stargardt disease | 0 probands | Single-strand conformational polymorphism–heteroduplex analysis sequencing and restriction fragment length polymorphism analysis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyango, O.; Mureithi, M.; Kithinji, D.; Jaoko, W.; Fujinami, K. Challenges and Opportunities in the Genetic Analysis of Inherited Retinal Dystrophies in Africa, a Literature Review. J. Pers. Med. 2023, 13, 239. https://doi.org/10.3390/jpm13020239
Onyango O, Mureithi M, Kithinji D, Jaoko W, Fujinami K. Challenges and Opportunities in the Genetic Analysis of Inherited Retinal Dystrophies in Africa, a Literature Review. Journal of Personalized Medicine. 2023; 13(2):239. https://doi.org/10.3390/jpm13020239
Chicago/Turabian StyleOnyango, Oscar, Marianne Mureithi, Dennis Kithinji, Walter Jaoko, and Kaoru Fujinami. 2023. "Challenges and Opportunities in the Genetic Analysis of Inherited Retinal Dystrophies in Africa, a Literature Review" Journal of Personalized Medicine 13, no. 2: 239. https://doi.org/10.3390/jpm13020239
APA StyleOnyango, O., Mureithi, M., Kithinji, D., Jaoko, W., & Fujinami, K. (2023). Challenges and Opportunities in the Genetic Analysis of Inherited Retinal Dystrophies in Africa, a Literature Review. Journal of Personalized Medicine, 13(2), 239. https://doi.org/10.3390/jpm13020239