
An Update on Tamoxifen and the Chemo-Preventive Potential of Vitamin E in Breast Cancer Management

 , , , ,
, , , ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Literature Search
3. A Brief Description of Tamoxifen and Its Interaction with the ER
4. Insight to the Chemo-Preventive Action of Tamoxifen in Breast Cancer
5. Acquired Resistance to Tamoxifen
6. Side Effects of Tamoxifen
7. Insight into Vitamin E in Breast Cancer Chemo-Prevention
8. Tamoxifen and Vitamin E Inhibitory Effects
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Miller, K.D.; Goding, S.A.; Jemal, A.; Siegel, R.L. Breast cancer statistics 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics: 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ismaila, N.; Allison, K.H.; Barlow, W.E.; Collyar, D.E.; Damodaran, S.; Henry, N.L.; Jhaveri, K.; Kalinsky, K.; Kuderer, N.M.; et al. Biomarkers for Adjuvant Endocrine and Chemotherapy in Early-Stage Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. 2022, 40, 1816–1837. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S. Triple-negative breast cancer: Epidemiology and management options. Drugs 2010, 70, 2247–2258. [Google Scholar] [CrossRef]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; Wang, Y.C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomized trials. Lancet 2011, 27, 771–778. [Google Scholar] [CrossRef]
- Lakhani, S.; Ellis, I.; Schnitt, S.; Schnitt, S.J.; Tan, P.H.; van de Vijver, M.J. WHO Classification of Tumors of the Breast, 4th ed.; IARC Press: Lyon, France, 2012; ISBN 978-92-832-2433-4. [Google Scholar]
- Rosenberg, S.M.; Partridge, A.H. Management of Breast Cancer in Very Young Women. Breast 2015, 24, S154–S158. [Google Scholar] [CrossRef]
- Kantor, E.D.; Rehm, C.D.; Du, M.; White, E.; Giovannucci, E.L. Trends in dietary supplement use among US adults from 1999–2012. JAMA 2016, 316, 1464–1474. [Google Scholar] [CrossRef]
- D’Andrea, G.M. Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J. Clin. 2005, 55, 319–321. [Google Scholar] [CrossRef]
- Hardy, M.L. Dietary supplement use in cancer care: Help or harm. Hematol. Oncol. Clin. N. Am. 2008, 22, 581–617. [Google Scholar] [CrossRef]
- Podszun, M.; Frank, J. Vitamin E-drug interactions: Molecular basis and clinical relevance. Nutr. Res. Rev. 2014, 27, 215–231. [Google Scholar] [CrossRef]
- Chmölz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 26, 14–43. [Google Scholar] [CrossRef]
- Galmés, S.; Serra, F.; Palou, A. Vitamin E Metabolic Effects and Genetic Variants: A Challenge for Precision Nutrition in Obesity and Associated Disturbances. Nutrients 2018, 10, 1919. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Saji, S.; Hirose, M.; Toi, M. Clinical significance of estrogen receptor in breast cancer. Cancer Chemother. Pharmacol. 2005, 1, 21–26. [Google Scholar] [CrossRef]
- Maximov, P.Y.; Lee, T.M.; Jordan, V.C. Discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr. Clin. Pharmacol. 2013, 8, 135–155. [Google Scholar] [CrossRef]
- Torchia, J.; Rose, D.W.; Inostroza, J.; Kamei, Y.; Westin, S.; Glass, C.K.; Rosenfeld, M.G. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997, 387, 677–684. [Google Scholar] [CrossRef]
- Musa, M.; Khan, M.O.; Copperwood, J.S. Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs). Curr. Med. Chem. 2007, 14, 1249–1261. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.K.; et al. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin. Cancer Res. 2008, 14, 5228–5235. [Google Scholar] [CrossRef]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Anticancer and drug resistance mechanisms. Muta. Res. 2005, 591, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Collins, M.M.; Rowsby, L.; Prestwich, G.A. monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J. Endocrinol. 1977, 75, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Dehal, S.S.; Kupfer, D. CYP2D6 catalyzes tamoxifen 4-hydroxylation in human liver. Cancer Res. 1997, 57, 3402–3406. [Google Scholar] [PubMed]
- Johnson, M.D.; Zuo, H.; Lee, K.H. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res. Treat. 2004, 85, 151–159. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Pike, A.C.; Brzozowski, A.M.; Hubbard, R.E.; Bonn, T.; Thorsell, A.G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999, 18, 4608–4618. [Google Scholar] [CrossRef]
- Pike, A.C.; Brzozowski, A.M.; Walton, J.; Hubbard, R.E.; Thorsell, A.G.; Li, Y.L.; Gustafsson, J.A.; Carlquist, M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.A. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Rosano, C.; Stec-Martyna, E.; Lappano, R.; Maggiolini, M. A structure-based approach for the discovery of new selective estrogen receptor modulators. Curr. Med. Chem. 2011, 18, 1188–1194. [Google Scholar] [CrossRef]
- Webb, P.; Lopez, G.N.; Uht, R.M.; Kusher, P.J. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell specific estrogen-like effects of antiestrogen. Mol. Endocrinol. 1995, 9, 443–456. [Google Scholar] [CrossRef]
- Clarke, R.B.; Anderson, E.; Howell, A. Steroid receptors in human breast cancer. Trends Endocrinol. Metab. 2004, 15, 316–323. [Google Scholar] [CrossRef]
- Fox, E.M.; Andrade, J.; Shupnik, M.A. Novel actions of estrogen to promote proliferation: Integration of cytoplasmic and nuclear pathways. Steroids 2009, 74, 622–627. [Google Scholar] [CrossRef]
- Doisneau-Sixou, S.F.; Sergio, C.M.; Carroll, J.S.; Hui, R.; Musgrove, E.A.; Sutherland, R.L. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186. [Google Scholar] [CrossRef]
- Peyrat, J.P.; Bonneterre, J. Type 1 IGF receptor in human breast diseases. Breast Cancer Res. Treat. 1992, 22, 59–67. [Google Scholar] [CrossRef]
- Baer-Dubowska, W.; Ignatowicz, E. Chemoprevention of Cancer Basic Mechanisms and Molecular Targets. In Carcinogenic and Anticarcinogenic food Components; Baer Dubowska, W., Bartoszek, A., Malejka-Giganti, D., Eds.; CRC Taylor&Francis Group: Boca Raton, FL, USA; London, UK, 2006; pp. 177–196. ISBN 978-0849320965. [Google Scholar]
- Nelson, H.D.; Fu, R.; Griffin, J.C.; Nygren, P.; Smith, M.E.; Humphrey, L. Systematic review: Comparative effectiveness of medications to reduce risk for primary breast cancer. Ann. Intern. Med. 2009, 151, 703–715. [Google Scholar] [CrossRef]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef]
- Coombes, R.C.; Hall, E.; Gibson, L.J.; Paridaens, R.; Jassem, J.; Delozier, T.; Jones, S.E.; Alvarez, I.; Bertelli, G.; Ortmann, O.; et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N. Engl. J. Med. 2004, 350, 1081–1092. [Google Scholar] [CrossRef]
- Cuzick, J.; Forbes, J.; Edwards, R. First results from the International Breast Cancer Intervention Study (IBIS-I): A randomised prevention trial. Lancet 2002, 360, 817–824. [Google Scholar] [CrossRef]
- Cuzick, J.; Powles, T.; Veronesi, U.; Forbes, J.; Edwards, R.; Ashley, S.; Boyle, P. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003, 361, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Forbes, J.F.; Sestak, I.; Cawthorn, S.; Hamed, H.; Holli, K.; Howell, A. Long-term results of tamoxifen prophylaxis for breast cancer—96-month follow-up of the randomized IBIS-I trial. J. Natl. Cancer Inst. 2007, 99, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Swaby, R.F.; Sharma, C.G.; Jordan, V.C. SERMs for the treatment and prevention of breast cancer. Rev. Endocr. Metab. Disord. 2007, 8, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Alencar, V.H.M.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptorpositive breast cancer ATLAS a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Wilkes, G.M.; Barton-Burke, M. 2020–2021 Oncology Nursing Drug Handbook; Jones & Bartlett Learning: Burlington, NJ, USA, 2019; ISBN 13: 978-1284171327. [Google Scholar]
- Parton, M.; Smith, I.E. Controversies in the management of patients with Breast cancer: Adjuvant endocrine therapy in premenopausal women. J. Clin. Oncol. 2008, 26, 745–752. [Google Scholar] [CrossRef]
- Kramer, R.; Brown, P. Should tamoxifen be used in breast cancer prevention? Drug Saf. 2004, 27, 979–989. [Google Scholar] [CrossRef]
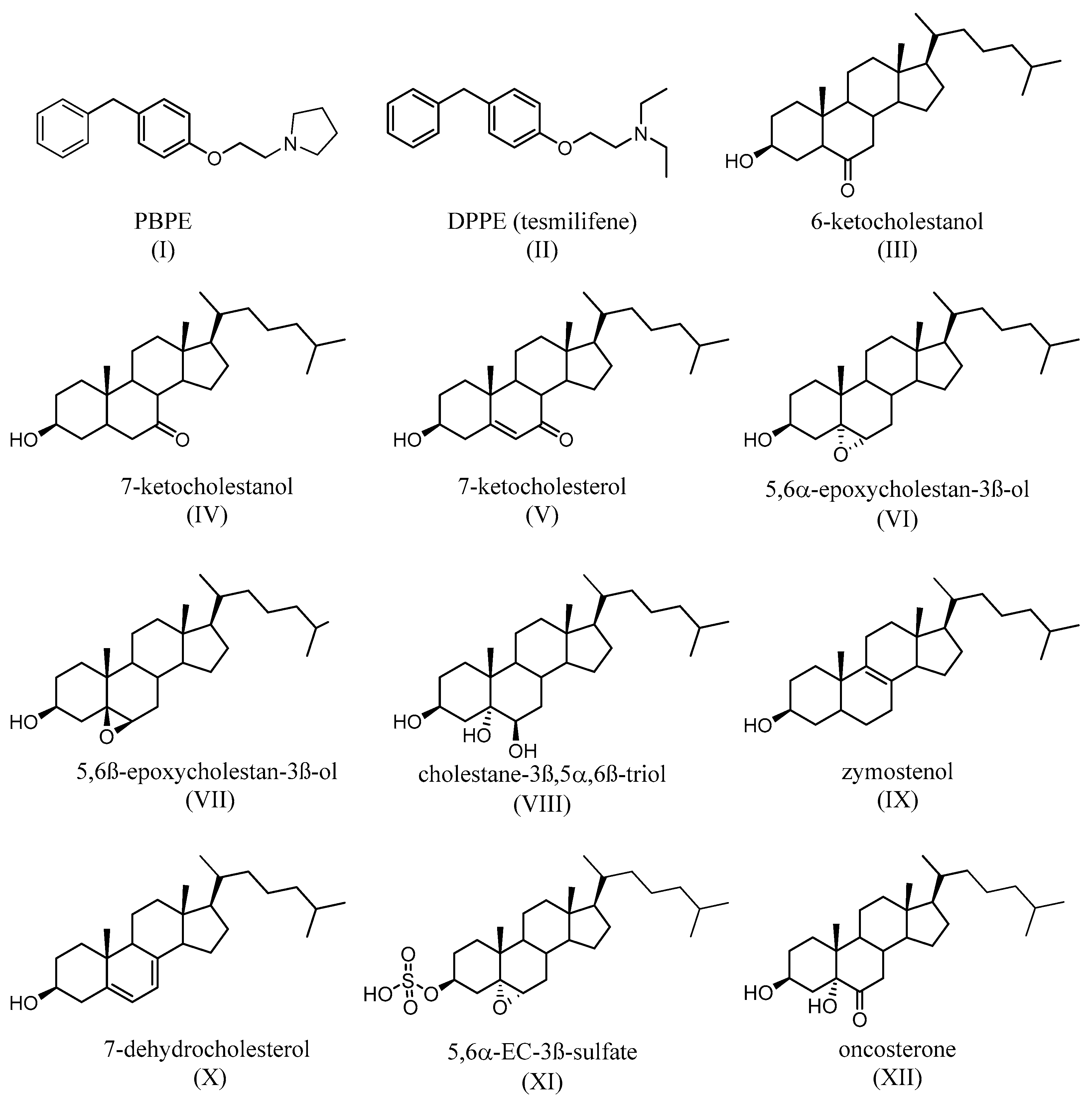
- de Médina, P.; Favre, G.; Poirot, M. Multiple targeting by the antitumor drug tamoxifen: A structure-activity study. Curr. Med. Chem. Anti-Cancer Agents. 2004, 4, 491–508. [Google Scholar] [CrossRef]
- Leignadier, J.; Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem. Pharmacol. 2017, 144, 18–28. [Google Scholar] [CrossRef]
- Bogush, T.A.; Polezhaev, B.B.; Mamichev, I.A.; Bogush, E.A.; Polotsky, B.E.; Tjulandin, S.A.; Ryabov, A.B. Tamoxifen Never Ceases to Amaze: New Findings on Non-Estrogen Receptor Molecular Targets and Mediated Effects. Cancer Investig. 2018, 36, 211–220. [Google Scholar] [CrossRef]
- Kedjouar, B.; Daunes, S.; Vilner, B.J.; Bowen, W.D.; Klaebe, A.; Faye, J.C.; Poirot, M. Structural similitudes between cytotoxic antiestrogen-binding site (AEBS) ligands and cytotoxic sigma receptor ligands. Evidence for a relationship between cytotoxicity and affinity for AEBS or sigma-2 receptor but not for sigma-1 receptor. Biochem. Pharmacol. 1999, 58, 1927–1939. [Google Scholar] [CrossRef]
- Kedjouar, B.; de Médina, P.; Oulad-Abdelghani, M.; Payré, B.; Silvente-Poirot, S.; Favre, G.; Faye, J.C.; Poirot, M. Molecular characterization of the microsomal tamoxifen binding site. J. Biol. Chem. 2004, 279, 34048–34061. [Google Scholar] [CrossRef]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cholesterol epoxide hydrolase and cancer. Curr. Opin. Pharmacol. 2012, 6, 696–703. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cancer Cholesterol and cancer in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef]
- Voisin, M.; de Medina, P.; Mallinger, A.; Dalenc, F.; Huc-Claustre, E.; Leignadier, J.; Serhan, N.; Soules, R.; Segale, G.; Mougel, A.; et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E9346–E9355. [Google Scholar] [CrossRef]
- de Medina, P.; Diallo, K.; Huc-Claustre, E.; Attia, M.; Soulès, R.; Silvente-Poirot, S.; Poirot, M. The 5,6-epoxycholesterol metabolic pathway in breast cancer: Emergence of new pharmacological targets. Br. J. Pharmacol. 2021, 178, 3248–3260. [Google Scholar] [CrossRef]
- Poirot, M.; Mallinger, A.; Dalenc, F.; Soulès, R.; Silvente-Poirot, S. Chemistry, biochemistry, metabolic fate and mechanism of action of 6-oxo-cholestan-3β,5α-diol (OCDO), a tumor promoter and cholesterol metabolite. Biochimie 2018, 153, 139–149. [Google Scholar] [CrossRef]
- Segala, G.; de Medina, P.; Iuliano, L.; Zerbinati, C.; Paillasse, M.R.; Noguer, E.; Dalenc, F.; Payré, B.; Jordan, V.C.; Record, M.; et al. 5,6-Epoxy-cholesterols contribute to the anticancer pharmacology of tamoxifen in breast cancer cells. Biochem. Pharmacol. 2013, 86, 175–189. [Google Scholar] [CrossRef]
- Aneja, R.; Zhou, J.; Zhou, B.F.; Chandra, R.; Joshi, H.C. Treatment of hormone-refractory breast cancer: Apoptosis and regression of human tumors implanted in mice. Mol. Cancer Ther. 2006, 5, 2366–2377. [Google Scholar] [CrossRef]
- Nielsen, K.V.; Ejlertsen, B.; Müller, S.; Møller, S.; Rasmussen, B.B.; Balslev, E.; Lænkholm, A.V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 might predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 345–355. [Google Scholar] [CrossRef]
- Rodriguez, D.; Ramkairsingh, M.; Lin, X.; Kapoor, A.; Major, P.; Tang, D. The central contributions of breast cancer stem cells in developing resistance to endocrine therapy in Estrogen Receptor (ER)-positive breast cancer. Cancers 2019, 11, 1028. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Houghton, J.; Iden, C.; Salter, J.; Farndon, J.; A’Hern, R.; Sainsbury, R.; Baum, M. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according estrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann. Oncol. 2006, 17, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, H.M.; Cheng, S.B.; Christensen, E.M.; Filardo, E.J. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol. Cell Endocrinol. 2015, 418, 207–219. [Google Scholar] [CrossRef]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef]
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.; Maggiolini, M.; et al. Tamoxifen through GPER upregulates aromatase expression: A novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 2014, 146, 273–285. [Google Scholar] [CrossRef]
- Arias-Pulido, H.; Royce, M.; Gong, Y.; Joste, N.; Lomo, L.; Lee, S.J.; Chaher, N.; Verschraegen, C.; Lara, J.; Prossnitz, E.R.; et al. GPR30 and estrogen receptor expression: New insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res. Treat. 2010, 123, 51–58. [Google Scholar] [CrossRef]
- Filardo, E.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef]
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell. Endocrinol. 2020, 506, 110762. [Google Scholar] [CrossRef]
- Butta, A.; MacLennan, K.; Flanders, K.C.; Sacks, N.P.; Smith, I.; McKinna, A.; Dowsett, M.; Wakefield, L.M.; Sporn, M.B.; Baum, M. Induction of transforming growth factor beta 1 in human breast cancer in vivo following tamoxifen treatment. Cancer Res. 1992, 52, 4261–4264. [Google Scholar]
- Brandt, S.; Kopp, A.; Grage, B.; Knabbe, C. Effects of tamoxifen on transcriptional level of transforming growth factor beta (TGF-beta) isoforms 1 and 2 in tumor tissue during primary treatment of patients with breast cancer. Anticancer Res. 2003, 23, 223–229. [Google Scholar]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331S–336S. [Google Scholar] [CrossRef]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008, 68, 5226–5235. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.; Cooke, T.G. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef]
- Meyer, D.S.; Bentires-Alj, M. Can phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibition ERase them all. Breast Cancer Res. 2010, 12, 315. [Google Scholar] [CrossRef]
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 2016, 50, 68–81. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer. Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef]
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.H.; Moustakas, A. Tamoxifen Inhibits TGF-β-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J. Cell Physiol. 2015, 230, 3084–3092. [Google Scholar] [CrossRef]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Rae, J.M. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response. Expert. Rev. Mol. Med. 2008, 10, e34. [Google Scholar] [CrossRef]
- Brauch, H.; Schwab, M. Prediction of tamoxifen outcome by genetic variation of CYP2D6 in post-menopausal women with early breast cancer. Br. J. Clin. Pharmacol. 2014, 77, 695–703. [Google Scholar] [CrossRef]
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.J.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; McLeod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2D6 and tamoxifen therapy. Clin. Pharmacol. Ther. 2018, 103, 770–777. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, S.; Duan, Y.; Ma, Y.; Wang, Y.; Ji, H.; Zhang, Q. GLUT1 participates in tamoxifen resistance in breast cancer cells through autophagy regulation Naunyn Schmiedebergs. Arch. Pharmacol. 2021, 394, 205–216. [Google Scholar] [CrossRef]
- Kang, S.S.; Chun, Y.K.; Hur, M.H.; Lee, H.K.; Kim, Y.J.; Hong, S.R.; Lee, J.H.; Lee, S.G.; Park, Y.K. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn. J. Cancer Res. 2002, 93, 1123–1128. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef]
- Jögi, A.; Ehinger, A.; Hartman, L.; Alkner, S. Expression of HIF-1α is related to a poor prognosis and tamoxifen resistance in contralateral breast cancer. PLoS ONE 2019, 14, e0226150. [Google Scholar] [CrossRef]
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1004–1018. [Google Scholar] [CrossRef]
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef]
- Tomková, V.; Sandoval-Acuña, C.; Torrealba, N.; Truksa, J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic. Biol. Med. 2019, 143, 510–521. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/ neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERα36+ breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef]
- Rajendran, S.; Swaroop, S.S.; Roy, J.; Inemai, E.; Murugan, S.; Rayala, S.K.; Venkatraman, G. p21 activated kinase-1 and tamoxifen—A deadly nexus impacting breast cancer outcomes. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188668. [Google Scholar] [CrossRef]
- Stender, J.D.; Nwachukwu, J.C.; Kastrati, I.; Kim, Y.; Strid, T.; Yakir, M.; Srinivasan, S.; Nowak, J.; Izard, T.; Rangarajan, E.S.; et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol. Cell. 2017, 65, 1122–1135.e5. [Google Scholar] [CrossRef]
- Saw, C.L.; Wu, Q.; Kong, A.N. Anti-cancer and potential chemopreventive actions of ginseng by activating Nrf2 (NFE2L2) anti-oxidative stress/anti-inflammatory pathways. Chin. Med. 2010, 5, 37. [Google Scholar] [CrossRef]
- Kwak, M.K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef]
- Kim, S.K.; Yang, J.W.; Kim, M.R.; Roh, S.H.; Kim, H.G.; Lee, K.Y.; Jeong, H.G.; Kang, K.W. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic. Biol. Med. 2008, 45, 537–546. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Wu, H.; Ru, P.; Hwang, L.; Trieu, V.; Mo, Y.Y. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 2011, 30, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef]
- Payré, B.; de Medina, P.; Boubekeur, N.; Mhamdi, L.; Bertrand-Michel, J.; Tercé, F.; Fourquaux, I.; Goudounèche, D.; Record, M.; Poirot, M.; et al. Microsomal antiestrogen-binding site ligands induce growth control and differentiation of human breast cancer cells through the modulation of cholesterol metabolism. Mol. Cancer Ther. 2008, 7, 3707–3718. [Google Scholar] [CrossRef]
- de Medina, P.; Silvente-Poirot, S.; Poirot, M. Tamoxifen and AEBS ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy 2009, 5, 1066–1067. [Google Scholar] [CrossRef]
- Pitroda, S.P.; Khodarev, N.N.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 5837–5841. [Google Scholar] [CrossRef]
- Maggiore, R.J.; Gross, C.P.; Hurria, A. Polypharmacy in older adults with cancer. Oncologist 2010, 15, 507–522. [Google Scholar] [CrossRef]
- Murphy, C.C.; Bartholomew, L.K.; Carpentier, M.Y.; Bluethmann, S.M.; Vernon, S.W. Adherence to adjuvant hormonal therapy among breast cancer survivors in clinical practice: A systematic review. Breast Cancer Res. Treat. 2012, 134, 459–478. [Google Scholar] [CrossRef]
- Day, R.; National Surgical Adjuvant Breast and Bowel Projet P-1 study (NSABP-1). Quality of life and tamoxifen in a breast cancer prevention trial: A summary of findings from the NSABP P-1 study. National Surgical Adjuvant Breast and Bowel Project. Ann. N. Y. Acad. Sci. 2001, 949, 143–150. [Google Scholar] [CrossRef]
- Polin, S.; Ascher, S. The effect of tamoxifen on the genital tract. Cancer Imaging 2008, 8, 135–145. [Google Scholar] [CrossRef]
- Braithwaite, R.S.; Chlebowski, R.T.; Lau, J.; George, S.; Hess, R.; Col, N.F. Meta-analysis of vascular and neoplastic events associated with tamoxifen. J. Gen. Intern. Med. 2003, 18, 937–947. [Google Scholar] [CrossRef]
- AlZaabi, A.; AlAmri, H.; ALAjmi, G.; Allawati, M.; Muhanna, F.; Alabri, R.; AlBusaidi, F.; AlGhafri, S.; Al-Mirza, A.A.; Al Baimani, K. Endometrial Surveillance in Tamoxifen and Letrozole Treated Breast Cancer Patients. Cureus 2021, 13, e20030. [Google Scholar] [CrossRef]
- Palmer, J.L.; Trotter, T.; Joy, A.A.; Carlson, L.E. Cognitive effects of tamoxifen in pre-menopausal women with breast cancer compared to healthy controls. J. Cancer Surviv. 2008, 2, 275–282. [Google Scholar] [CrossRef]
- Espeland, M.A.; Shumaker, S.A.; Limacher, M.; Rapp, S.R.; Bevers, T.B.; Barad, D.H.; Coker, L.H.; Gaussoin, S.A.; Stefanick, M.L.; Lane, D.S.; et al. Relative effects of tamoxifen raloxifene and conjugated equine estrogens on cognition. J. Womens Health 2010, 19, 371–379. [Google Scholar] [CrossRef]
- Lee, K.C.; Ray, G.T.; Hunkeler, E.M.; Finley, P.R. Tamoxifen treatment and new-onset depression in breast cancer patients. Psychosomatics 2007, 48, 205–210. [Google Scholar] [CrossRef]
- Blencowe, N.S.; Reichl, C.; Gahir, J.; Paterson, I. The use of Nolvadex in the treatment of generic Tamoxifen-associated small joint arthralgia. Breast 2010, 19, 243–245. [Google Scholar] [CrossRef]
- Alekshun, T.; Patterson, S. Management of hot flashes in men with prostate cancer treated with androgen deprivation therapy. Support Cancer Ther. 2006, 4, 30–37. [Google Scholar] [CrossRef]
- Perez, E.A. Safety profiles of tamoxifen and aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Ann. Oncol. 2007, 18, 26–35. [Google Scholar] [CrossRef]
- Avis, N.E. Breast cancer survivors and hot flashes: The search for nonhormonal treatments. J. Clin. Oncol. 2008, 26, 5008–5010. [Google Scholar] [CrossRef] [PubMed]
- Cella, D.; Fallowfield, L. Recognition and management of treatment-related side effects for breast cancer patients receiving adjuvant endocrine therapy. Breast Cancer Res. Treat. 2008, 107, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Morrow, P.K.; Mattair, D.N.; Hortobagyi, G.N. Hot flushes: A review of pathophysiology and treatment modalities. Oncologist 2011, 16, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.K.; Jacobs, R.L.; Vance, D.E. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology 2010, 52, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Jatobá, C.A.; Rezende, A.A.; Paiva Rodrigues, S.J.; Almeida Câmara, M.M.; das Graças Almeida, M.; Freire-Neto, F.; da Rocha, L.R.; da Medeiros, A.C.; Brandão-Neto, J.; Carvalho Formiga, M.C.; et al. Liver iron overload induced by tamoxifen in diabetic and non-diabetic female Wistar rats. Biometals 2008, 21, 171–178. [Google Scholar] [CrossRef]
- Nguyen, M.C.; Stewart, R.B.; Banerji, M.A.; Gordon, D.H.; Kral, J.G. Relationships between tamoxifen use liver fat and body fat distribution in women with breast cancer. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 296–298. [Google Scholar] [CrossRef]
- Nemoto, Y.; Saibara, T.; Ogawa, Y.; Zhang, T.; Xu, N.; Ono, M.; Akisawa, N.; Iwasaki, S.; Maeda, T.; Onishi, S. Tamoxifen-induced nonalcoholic steatohepatitis in breast cancer patients treated with adjuvant tamoxifen. Intern. Med. 2002, 41, 345–350. [Google Scholar] [CrossRef]
- Ohnishi, T.; Ogawa, Y.; Saibara, T.; Nishioka, A.; Kariya, S.; Fukumoto, M.; Onishi, S.; Yoshida, S. CYP17 polymorphism and tamoxifen-induced hepatic steatosis. Hepatol. Res. 2005, 33, 178–180. [Google Scholar] [CrossRef]
- Liu, C.L.; Yang, T.L. Sequential changes in serum triglyceride levels during adjuvant tamoxifen therapy in breast cancer patients and the Effect of dose reduction. Breast Cancer Res. Treat. 2003, 79, 11–16. [Google Scholar] [CrossRef]
- Kim, Y.A.; Lee, S.; Jung, J.W.; Kwon, Y.J.; Lee, G.B.; Shin, D.G.; Park, S.S.; Yun, J.; Jang, Y.S.; Cho, D.H. Severe acute pancreatitis due to tamoxifen-induced hypertriglyceridemia with diabetes mellitus. Chin. J. Cancer Res. 2014, 26, 341–344. [Google Scholar] [CrossRef]
- Baumgart, J.; Nilsson, K.; Evers, A.S.; Kallak, T.K.; Poromaa, I.S. Sexual dysfunction in women on adjuvant endocrine therapy after breast cancer. Menopause 2013, 20, 162–168. [Google Scholar] [CrossRef]
- Tevaarwerk, A.J.; Wang, M.; Zhao, F.; Fetting, J.H.; Cella, D.; Wagner, L.I.; Martino, S.; Ingle, J.N.; Sparano, J.A.; Solin, L.J.; et al. Phase III comparison of tamoxifen versus tamoxifen plus ovarian function suppression in premenopausal women with node-negative, hormone-receptor positive breast cancer: A trial of Eastern Cooperative Oncology Group. J. Clin. Oncol. 2014, 32, 3948–3958. [Google Scholar] [CrossRef]
- Carmassi, C.; Cordone, A.; Dell’Oste, V.; Pedrinelli, V.; Pardini, F.; Simoncini, M.; Dell’Osso, L. Prescribing Tamoxifen in Patients with Mood Disorders: A Systematic Review of Potential Antimanic Versus Depressive Effects. J. Clin. Psychopharmacol. 2021, 41, 450–460. [Google Scholar] [CrossRef]
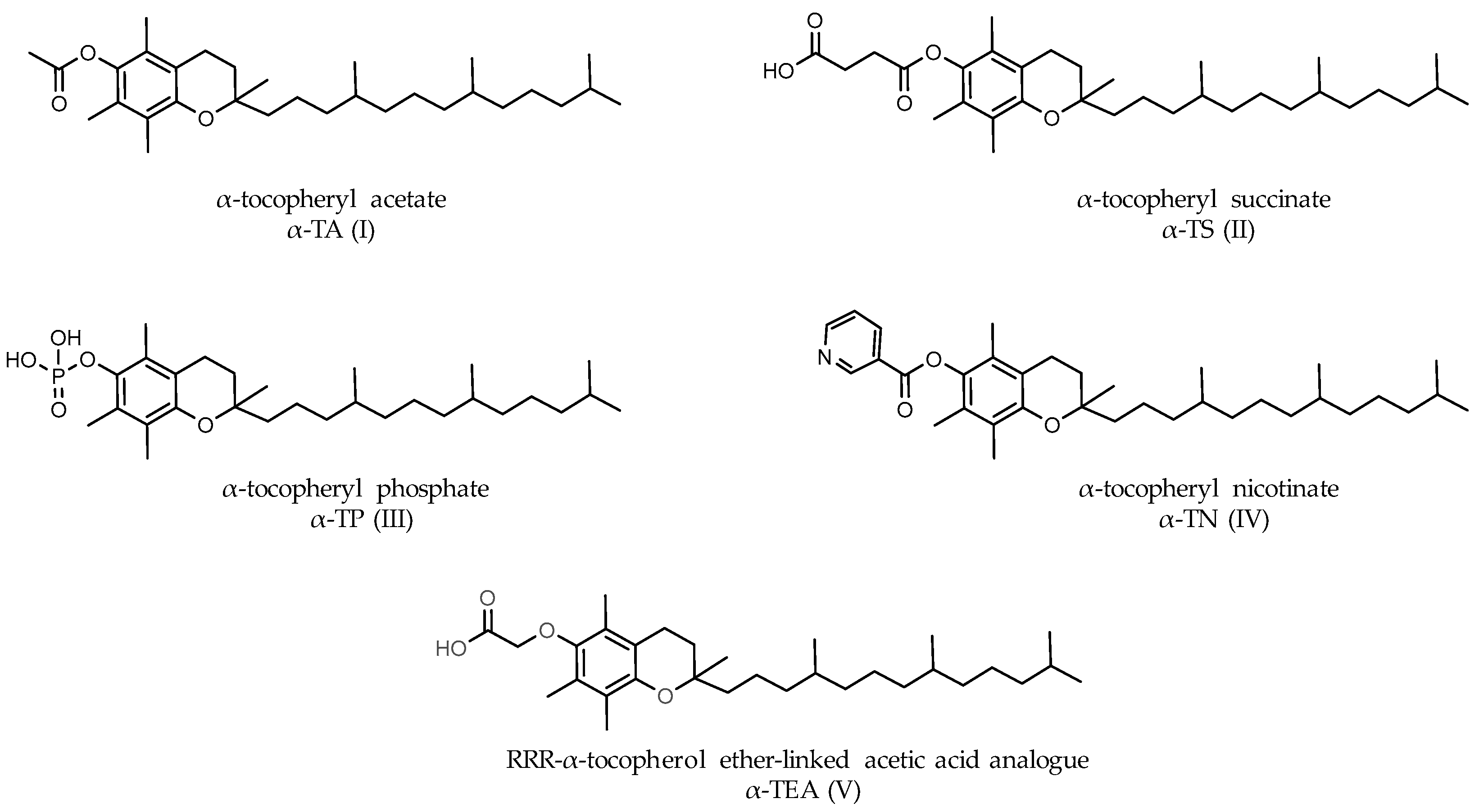
- Shah, S.; Sylvester, P.W. Tocotrienol-induced caspase-8 activation is unrelated to death receptor apoptotic signaling in neoplastic mammary epithelial cells. Exp. Biol. Med. 2004, 229, 745–755. [Google Scholar] [CrossRef]
- Nesaretnam, K.; Stephen, R.; Dils, R.; Darbre, P. Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids 1998, 33, 461–469. [Google Scholar] [CrossRef]
- Nesaretnam, K.; Selvaduray, K.R.; Abdul, R.G.; Veerasenan, S.D.; Gomez, P.A. Effectiveness of tocotrienol-rich fraction combined with tamoxifen in the management of women with early breast cancer: A pilot clinical trial. Breast Cancer Res. 2010, 12, R81. [Google Scholar] [CrossRef]
- Nesaretnam, K.; Meganathan, P.; Veerasenan, S.D.; Selvaduray, K.R. Tocotrienols and breast cancer: The evidence to date. Genes Nutr. 2012, 7, 3–9. [Google Scholar] [CrossRef]
- Shun, M.C.; Yu, W.; Gapor, A.; Parsons, R.; Atkinson, J.; Sanders, B.G.; Kline, K. Pro-apoptotic mechanisms of action of a novel vitamin E analog (α-TEA) and a naturally occurring form of vitamin E (delta-tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr. Cancer 2004, 48, 95–105. [Google Scholar] [CrossRef]
- Takahashi, K.; Loo, G. Disruption of mitochondria during tocotrienol-induced apoptosis in MDA-MB-231 human breast cancer cells. Biochem. Pharmacol. 2004, 67, 315–324. [Google Scholar] [CrossRef]
- Shah, S.J.; Sylvester, P.W. γ-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor κB activity. Exp. Biol. Med. 2005, 230, 235–241. [Google Scholar] [CrossRef]
- IARC. Tamoxifen in Some Pharmaceutical Drugs; IARC: Lyon, France, 1996; p. 253. [Google Scholar]
- Sylvester, P.W.; Shah, S.J. Mechanisms mediating the antiproliferative and apoptotic effects of vitamin E in mammary cancer cells. Front. Biosci. 2005, 10, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Comitato, R.; Nesaretnam, K.; Leoni, G.; Ambra, R.; Canali, R.; Bolli, A.; Marino, M.; Virgili, F. A novel mechanism of natural vitamin E tocotrienol activity: Involvement of ERbeta signal transduction. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E427–E437. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, C.; Hyatt, J.A.; Vraka, P.S.; Papas, A.; Papas, K.A.; Neophytou, C.; Hadjivassiliou, V.; Constantinou, A.I. Induction of caspase-independent programmed cell death by vitamin E natural homologs and synthetic derivatives. Nutr. Cancer 2009, 61, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Wali, V.B.; Bachawal, S.V.; Sylvester, P.W. Endoplasmic reticulum stress mediates gamma-tocotrienol-induced apoptosis in mammary tumor cells. Apoptosis 2009, 14, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Samant, G.V.; Wali, V.B.; Sylvester, P.W. Anti-proliferative effects of gammatocotrienol on mammary tumour cells are associated with suppression of cell cycle progression. Cell Prolif. 2010, 43, 77–83. [Google Scholar] [CrossRef]
- Park, S.K.; Sanders, B.G.; Kline, K. Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res. Treat. 2010, 124, 361–375. [Google Scholar] [CrossRef]
- Bachawal, S.V.; Wali, V.B.; Sylvester, P.W. Enhanced antiproliferative and apoptotic response to combined treatment of gamma-tocotrienol with erlotinib or gefitinib in mammary tumor cells. BMC Cancer 2010, 8, 84. [Google Scholar] [CrossRef]
- Wada, S. Cancer preventive effects of vitamin E. Curr. Pharm. Biotechnol. 2012, 13, 156–164. [Google Scholar] [CrossRef]
- Khallouki, F.; de Medina, P.; Caze-Subra, S.; Bystricky, K.; Balaguer, P.; Poirot, M.; Silvente-Poirot, S. Molecular and Biochemical Analysis of the Estrogenic and Proliferative Properties of Vitamin E Compounds. Front. Oncol. 2016, 5, 287. [Google Scholar] [CrossRef]
- Sailo, B.L.; Banik, K.; Padmavathi, G.; Javadi, M.; Bordoloi, D.; Kunnumakkara, A.B. Tocotrienols: The promising analogues of vitamin E for cancer therapeutics. Pharmacol. Res. 2018, 130, 259–272. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Krishnan, K.; Aggarwal, B.B. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J. Biol. Chem. 2007, 282, 809–820. [Google Scholar] [CrossRef]
- Loganathan, R.; Selvaduray, K.R.; Nesaretnan, K.; Radhakrishnan, A.K. Tocotrienols promote apoptosis in human breast cancer cells by inducing poly(ADP-ribose) polymerase cleavage and inhibiting nuclear factor kappa-B activity. Cell Prolif. 2013, 46, 203–213. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Elangovan, S.; Wu, J.M. Differential suppression of proliferation in MCF-7 and MDA-MB-231 breast cancer cells exposed to alpha-, gamma- and delta-tocotrienols is accompanied by altered expression of oxidative stress modulatory enzymes. Anticancer Res. 2010, 30, 4169–4176. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Sundaram, C.; Prasad, S.; Kannappan, R. Tocotrienols, the vitamin E of the 21st century: Its potential against cancer and other chronic diseases. Biochem. Pharmacol. 2010, 80, 1613–1631. [Google Scholar] [CrossRef]
- Elangovan, S.; Hsieh, T.C.; Wu, J.M. Growth inhibition of human MDA-mB-231 breast cancer cells by delta-tocotrienol is associated with loss of cyclin D1/ CDK4 expression and accompanying changes in the state of phosphorylation of the retinoblastoma tumor suppressor gene product. Anticancer Res. 2008, 28, 2641–2647. [Google Scholar]
- Selvaduray, K.R.; Radhakrishnan, A.; Kutty, M.K.; Nesaretnam, K. Palm tocotrienols inhibit proliferation of murine mammary cancer cells and induce expression of interleukin-24 mRNA. J. Interferon Cytokine Res. 2010, 30, 909–916. [Google Scholar] [CrossRef]
- Hirose, M.; Masuda, A.; Inoue, T.; Fukushima, S.; Ito, N. Modification by antioxidants and p,p ′-diaminodiphenylmethane of 7,12-dimethylbenz(α)anthracene-induced carcinogenesis of the mammary gland and ear duct in CD rats. Carcinogenesis 1986, 7, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Gould, M.N.; Haag, J.D.; Kennan, W.S.; Tanner, M.A.; Elson, C.E. A comparison of tocopherol and tocotrienol for the chemoprevention of chemically induced rat mammary tumors. Am. J. Clin. Nutr. 1991, 53, 1068S–1070S. [Google Scholar] [CrossRef]
- Iqbal, J.; Minhajuddin, M.; Beg, Z.H. Suppression of 7,12-dimethylbenz[alpha]anthracene-induced carcinogenesis and hypercholesterolaemia in rats by tocotrienol-rich fraction isolated from rice bran oil. Eur. J. Cancer Prev. 2003, 12, 447–453. [Google Scholar] [CrossRef]
- Suh, N.; Paul, S.; Lee, H.J.; Ji, Y.; Lee, M.J.; Yang, C.S.; Reddy, B.S.; Newmark, H.L. Mixed tocopherols inhibit N-methyl-N-nitrosourea-induced mammary tumor growth in rats. Nutr. Cancer 2007, 59, 76–81. [Google Scholar] [CrossRef]
- Smolarek, A.K.; Suh, N. Chemopreventive activity of vitamin E in breast cancer: A focus on gamma and delta-tocopherol. Nutrients 2011, 3, 962–986. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ju, J.; Paul, S.; So, J.Y.; DeCastro, A.; Smolarek, A.K.; Lee, M.J.; Yang, C.S.; Newmark, H.L.; Suh, N. Mixed tocopherols prevent mammary tumorigenesis by inhibiting estrogen action and activating PPAR-gamma. Clin. Cancer Res. 2009, 15, 4242–4249. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Luo, P.; Zeng, Z.; Wang, H.; Malafa, M.; Suh, N. Vitamin E and cancer prevention: Studies with different forms of tocopherols and tocotrienols. Mol. Carcinog. 2020, 59, 365–389. [Google Scholar] [CrossRef]
- Abuasal, B.S.; Qosa, H.P.; Sylvester, W.; Kaddoumi, B. Comparison of the intestinal absorption and bioavailability of gamma-tocotrienol and alpha-tocopherol: In vitro, in situ and in vivo studies. Biopharm. Drug Dispos. 2012, 33, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Abu-Fayyad, A.; Behery, F.; Sallam, A.A.; Alqahtani, S.; Ebrahim, H.; El Sayed, K.A.; Kaddoumi, A.; Sylvester, P.W.; Carroll, J.L.; Cardelli, J.A.; et al. PEGylated γ-tocotrienol isomer of vitamin E: Synthesis, characterization, in vitro cytotoxicity, and oral bioavailability. Eur. J. Pharm. Biopharm. 2015, 96, 185–195. [Google Scholar] [CrossRef]
- Tan, D.M.; Fu, J.Y.; Wong, F.S.; Er, H.M.; Chen, Y.S.; Nesaretnam, K. Tumor regression and modulation of gene expression via tumor-targeted tocotrienol niosomes. Nanomedicine 2017, 12, 2487–2502. [Google Scholar] [CrossRef]
- Pham, J.; Nayel, A.; Hoang, C.; Elbayoumi, T. Enhanced effectiveness of tocotrienol-based nano-emulsified system for topical delivery against skin carcinomas. Drug Deliv. 2016, 23, 1514–1524. [Google Scholar] [CrossRef]
- Anderson, K.; Simmons-Menchaca, M.; Lawson, K.A.; Atkinson, J.; Sanders, B.G.; Kline, K. Differential response of human ovarian cancer cells to induction of apoptosis by vitamin E Succinate and vitamin E analogue alpha-TEA. Cancer Res. 2004, 64, 4263–4269. [Google Scholar] [CrossRef]
- Neuzil, J.; Weber, T.; Schröder, A.; Lu, M.; Ostermann, G.; Gellert, N.; Mightne, G.C.; Olejnicka, B.; Nègre-Salvayre, A.; Stícha, M.; et al. Induction of cancer cell apoptosis by alpha-tocopheryl succinate: Molecular pathways and structural requirements. FASEB J. 2001, 15, 403–415. [Google Scholar] [CrossRef]
- Neuzil, J. Tocopheryl succinate epitomizes a compound with a shift in biological activity due to pro-vitamin-to-vitamin conversion. Biochem. Biophys. Res. Commun. 2002, 293, 1309–1313. [Google Scholar] [CrossRef]
- Prasad, K.; Kumar, B.; Yan, X.; Hanson, A.; Cole, W. Alpha-tocopheryl succinate, the most effective form of vitamin E for adjuvant cancer treatment: A review. J. Am. Coll. Nutr. 2003, 22, 108–117. [Google Scholar] [CrossRef]
- Kline, K.; Yu, W.; Sanders, B.G. Vitamin E and breast cancer. J. Nutr. 2004, 134, 3458S–3462S. [Google Scholar] [CrossRef]
- Zingg, J.M. Molecular and cellular activities of vitamin E analogues. Mini Rev. Med. Chem. 2007, 7, 543–558. [Google Scholar] [CrossRef]
- Tiwary, R.; Yu, W.; Li, J.; Park, S.K.; Sanders, B.G.; Kline, K. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS ONE 2010, 5, e11865. [Google Scholar] [CrossRef]
- Tiwary, R.; Yu, W.; Sanders, B.G.; Kline, K. α-TEA cooperates with MEK or mTOR inhibitors to induce apoptosis via targeting IRS/PI3K pathways. Br. J. Cancer 2011, 104, 101–109. [Google Scholar] [CrossRef]
- Hahn, T.; Szabo, L.; Gold, M.; Ramanathapuram, L.; Hurley, L.H.; Akporiaye, E.T. Dietary administration of the proapoptotic vitamin E analogue α-tocopheryl-oxyacetic acid inhibits metastatic murine breast cancer. Cancer Res. 2006, 66, 9374–9378. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.; Jagadish, B.; Mash, E.A.; Garrison, K.; Akporiaye, E.T. α- Tocopheryloxyacetic acid: A novel chemotherapeutic that stimulates the antitumor immune response. Breast Cancer Res. 2011, 13, R4. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Shun, M.C.; Anderson, K.; Chen, H.; Sanders, B.G.; Kline, K. α-TEA inhibits survival and enhances death pathways in cisplatin sensitive and resistant human ovarian cancer cells. Apoptosis 2006, 11, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yu, W.; Hu, Z.; Jia, L.; Iyer, V.R.; Sanders, B.G.; Kline, K. Involvement of JNK/p73/NOXA in vitamin E analog-induced apoptosis of human breast cancer cells. Mol. Carcinog. 2008, 47, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J. Vitamin E succinate and cancer treatment: A vitamin E prototype for selective antitumor activity. Br. J. Cancer 2003, 89, 1822–1826. [Google Scholar] [CrossRef]
- Kozin, S.V.; Shkarin, P.; Gerweck, L.E. The cell transmembrane pH gradient in tumors enhances cytotoxicity of specific weak acid chemotherapeutics. Cancer Res. 2001, 61, 4740–4743. [Google Scholar]
- Neuzil, J.; Tomasetti, M.; Mellick, A.S.; Alleva, R.; Salvatore, B.A.; Birringer, M.; Fariss, M.W. Vitamin E analogues: A new class of inducers of apoptosis with selective anti-cancer effects. Curr. Cancer Drug Targets 2004, 4, 355–372. [Google Scholar] [CrossRef]
- Yu, W.; Sanders, B.G.; Kline, K. RRR-alpha-tocopheryl succinate induction of DNA synthesis arrest of human MDA-MB-435 cells involves TGF-beta-independent activation of p21Waf1/Cip1. Nutr. Cancer 2002, 43, 227–236. [Google Scholar] [CrossRef]
- Constantinou, C.; Papas, A.; Constantinou, A. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int. J. Cancer 2008, 123, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Malafa, M.P.; Fokum, F.D.; Smith, L.; Louis, A. Inhibition of angiogenesis and promotion of melanoma dormancy by vitamin E succinate. Ann. Surg. Oncol. 2002, 9, 1023–1032. [Google Scholar] [CrossRef]
- Yu, W.; Tiwary, R.; Li, J.; Park, S.K.; Jia, L.; Xiong, A.; Simmons-Menchaca, M.; Sanders, B.G.; Kline, K. α-TEA induces apoptosis of human breast cancer cells via activation of TRAIL/DR5 death receptor pathway. Mol. Carcinog. 2010, 49, 964–973. [Google Scholar] [CrossRef]
- Schindler, R.; Mentlein, R. Flavonoids and vitamin E reduce the release of the angiogenic peptide vascular endothelial growth factor from human tumor cells. J. Nutr. 2006, 136, 1477–1482. [Google Scholar] [CrossRef]
- Kline, K.; Lawson, K.A.; Yu, W.; Sanders, B.G. Vitamin E and cancer. Vitam. Horm. 2007, 76, 435–461. [Google Scholar] [CrossRef]
- Dalen, H.; Neuzil, J. Alpha-tocopheryl succinate sensitises a T lymphoma cell line to TRAIL-induced apoptosis by suppressing NF-kappaB activation. Br. J. Cancer 2003, 88, 153–158. [Google Scholar] [CrossRef]
- Neuzil, J.; Wang, X.F.; Dong, L.F.; Low, P.; Ralph, S.J. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006, 580, 5125–5129. [Google Scholar] [CrossRef]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Freeman, R.; Liu, J.; Zobalova, R.; Marin-Hernandez, A.; Stantic, M.; Rohlena, J.; Rodriguez-Enriquez, S.; Valis, K.; Butcher, B.; et al. Suppression of tumor growth in vivo by the mitocan α-tocopheryl succinate requires respiratory complex II. Clin. Cancer Res. 2009, 15, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Jameson, V.J.A.; Tilly, D.; Cerny, J.; Mahdavian, E.; Marín-Hernández, A.; Hernández-Esquivel, L.; Rodríguez-Enríquez, S.; Stursa, J.; Witting, P.K.; et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anticancer activity via mitochondrial complex II. J. Biol. Chem. 2011, 286, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Rohlena, J.; Dong, L.F.; Kluckova, K.; Zobalova, R.; Goodwin, J.; Tilly, D.; Stursa, J.; Pecinova, A.; Philimonenko, A.; Hozak, P.; et al. Mitochondrially targeted α-tocopheryl succinate is antiangiogenic: Potential benefit against tumor angiogenesis but caution against wound healing. Antioxid. Redox Signal. 2011, 15, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Low, P.; Dyason, J.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J. α-Tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4333. [Google Scholar] [CrossRef]
- Hama, S.; Utsumi, S.; Fukuda, Y.; Nakayama, K.; Okamura, Y. Development of a novel drug delivery system consisting of an antitumor agent tocopheryl succinate. J. Control. Release 2012, 161, 843–851. [Google Scholar] [CrossRef]
- Youk, H.; Lee, E.; Choi, M.; Lee, Y.; Chung, J.H. Enhanced anticancer efficacy of α-tocopheryl succinate by conjugation with polyethylene glycol. J. Control. Release 2005, 107, 43–52. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2008, 16, CD007176. [Google Scholar] [CrossRef]
- Potter, J.D. The failure of cancer chemoprevention. Carcinogenesis 2014, 35, 974–982. [Google Scholar] [CrossRef]
- Dorjgochoo, T.; Shrubsole, M.J.; Shu, X.O.; Lu, W.; Ruan, Z.; Zheng, Y.; Cai, H.; Dai, Q.; Gu, K.; Gao, Y.T.; et al. Vitamin supplement use and risk for breast cancer: The Shanghai Breast Cancer Study. Breast Cancer Res. Treat. 2008, 111, 269–278. [Google Scholar] [CrossRef]
- Sharhar, S.; Normah, H.; Fatimah, A.; Fadilah, R.N.; Rohi, G.A.; Amin, I.; Cham, B.G.; Rizal, R.M.; Fairulnizal, M.N. Antioxidant intake and status, and oxidative stress in relation to breast cancer risk: A case-control study. Asian Pac. J. Cancer Prev. 2008, 9, 343–349. [Google Scholar]
- Nechuta, S.; Lu, W.; Chen, Z.; Zheng, Y.; Gu, K.; Cai, H.; Zheng, W.; Shu, X.O. Vitamin supplement use during breast cancer treatment and survival: A prospective cohort study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 262–271. [Google Scholar] [CrossRef]
- Pan, S.Y.; Zhou, J.; Gibbons, L.; Morrison, H.; Wen, S.W. Antioxidants and breast cancer risk—A population-based case-control study in Canada. BMC Cancer 2011, 11, 372. [Google Scholar] [CrossRef]
- Adzersen, K.H.; Jess, P.; Freivogel, K.W.; Gerhard, I.; Bastert, G. Raw and cooked vegetables, fruits, selected micronutrients, and breast cancer risk: A case-control study in Germany. Nutr. Cancer 2003, 46, 131–137. [Google Scholar] [CrossRef]
- Nissen, S.B.; Tjonneland, A.; Stripp, C.; Olsen, A.; Christensen, J.; Overvad, K.; Dragsted, L.O.; Thomsen, B. Intake of vitamins A, C, and E from diet and supplements and breast cancer in postmenopausal women. Cancer Causes Control 2003, 14, 695–704. [Google Scholar] [CrossRef]
- Zaroukian, S.; Pineault, R.; Gandini, S.; Lacroix, A.; Ghadirian, P. Correlation between nutritional biomarkers and breast cancer: A case-control study. Breast 2005, 14, 209–223. [Google Scholar] [CrossRef]
- Fink, B.N.; Gaudet, M.M.; Britton, J.A.; Abrahamson, P.E.; Teitelbaum, S.L.; Jacobson, J.; Bell, P.; Thomas, J.A.; Kabat, G.C.; Neugut, A.I.; et al. Fruits, vegetables, and micronutrient intake in relation to breast cancer survival. Breast Cancer Res. Treat. 2006, 98, 199–208. [Google Scholar] [CrossRef]
- Cui, Y.; Shikany, J.M.; Liu, S.; Shagufta, Y.; Rohan, T.E. Selected antioxidants and risk of hormone receptor-defined invasive breast cancers among postmenopausal women in the Women’s Health Initiative Observational Study. Am. J. Clin. Nutr. 2008, 87, 1009–1018. [Google Scholar] [CrossRef]
- Ishitani, K.; Lin, J.; Manson, J.E.; Buring, J.E.; Zhang, S.M. A prospective study of multivitamin supplement use and risk of breast cancer. Am. J. Epidemiol. 2008, 167, 1197–1206. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and metaanalysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nagorni, A.; Nikolova, D. Meta-analysis: Antioxidant supplements for primary and secondary prevention of colorectal adenoma. Aliment. Pharmacol. Ther. 2006, 24, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, K.; Stampfer, M. Vitamin and mineral supplements for cancer prevention: Issues and evidence. Am. J. Clin. Nutr. 2007, 85, 289S–292S. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.N.; Rajput, S.; Dey, K.K.; Parekh, A.; Das, S.; Mazumdar, A.; Mandal, M. Celecoxib alleviates tamoxifen-instigated angiogenic effects by ROS-dependent VEGF/VEGFR2 autocrine signaling. BMC Cancer 2013, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaëth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Han, N.N.; Zhou, Q.; Huang, Q.; Liu, K.J. Carnosic acid cooperates with tamoxifen to induce apoptosis associated with caspase-3 activation in breast cancer cells in vitro and in vivo. Biomed. Pharmacother. 2017, 89, 827–837. [Google Scholar] [CrossRef]
- Charalambous, C.; Pitta, C.A.; Constantinou, A.I. Equol enhances tamoxifen’s anti-tumor activity by induction of caspase-mediated apoptosis in MCF-7 breast cancer cells. BMC Cancer 2013, 13, 238. [Google Scholar] [CrossRef]
- Jena, S.K.; Suresh, S.; Sangamwar, A.T. Modulation of tamoxifen-induced hepatotoxicity by tamoxifen-phospholipid complex. J. Pharm. Pharmacol. 2015, 67, 1198–1206. [Google Scholar] [CrossRef]
- Pandey, S.K.; Ghosh, S.; Maiti, P.; Haldar, C. Therapeutic efcacy and toxicity of tamoxifen loaded PLA nanoparticles for breast cancer. Int. J. Biol. Macromol. 2015, 72, 309–319. [Google Scholar] [CrossRef]
- Ganji-Harsini, S.; Khazaei, M.; Rashidi, Z.; Ghanbari, A. Thymoquinone Could Increase the Efficacy of Tamoxifen Induced Apoptosis in Human Breast Cancer Cells: An In Vitro Study. Cell J. 2016, 18, 245–254. [Google Scholar] [CrossRef]
- Motawi, T.K.; Abdelazim, S.A.; Darwish, H.A.; Elbaz, E.M.; Shouman, S.A. Modulation of Tamoxifen Cytotoxicity by Caffeic Acid Phenethyl Ester in MCF-7 Breast Cancer Cells. Oxid. Med. Cell Longev. 2016, 2016, 3017108. [Google Scholar] [CrossRef]
- Catanzaro, E.; Seghetti, F.; Calcabrini, C.; Rampa, A.; Gobbi, S.; Sestili, P.; Turrini, E.; Maffei, F.; Hrelia, P.; Bisi, A.; et al. Identification of a new tamoxifen-xanthene hybrid as pro-apoptotic anticancer agent. Bioorg. Chem. 2019, 86, 538–549. [Google Scholar] [CrossRef]
- Adikwu, E.; Ebinyo, N.C.; Benalayefa, O. Protective effect of lycopene against tamoxifen-induced hepatotoxicity in albino rats. Biomed. Biotechnol. Res. J. 2020, 4, 69. [Google Scholar]
- Ghatreh, K.S.; Farrokhi, E.; Tabatabaee, A.; Jalilian, N.; Jafari, M. Synergistic effects of lauryl gallate and tamoxifen on human breast cancer cell. Iran J. Public Health 2020, 49, 1324–1329. [Google Scholar] [CrossRef]
- Famurewa, A.C.; Ekeleme-Egedigwe, C.A.; David, E.E.; Eleazu, C.O.; Folawiyo, A.M.; Obasi, N.A. Zinc abrogates anticancer drug tamoxifen-induced hepatotoxicity by suppressing redox imbalance, NO/iNOS/NF-ĸB signaling, and caspase-3-dependent apoptosis in female rats. Toxicol. Mech. Methods 2020, 30, 115–123. [Google Scholar] [CrossRef]
- Yen, C.; Zhao, F.; Yu, Z.; Zhu, X.; Li, C.G. Interactions Between Natural Products and Tamoxifen in Breast Cancer: A Comprehensive Literature Review. Front. Pharmacol. 2022, 13, 847113. [Google Scholar] [CrossRef]
- Yuvaraj, S.; Premkumar, V.G.; Vijayasarathy, K.; Gangadaran, S.G.D.; Sachdanandam, P. Augmented antioxidant status in tamoxifen treated postmenopausal women with breast cancer on co-administration with coenzyme Q 10, niacin and riboflavin. Cancer Chemother. Pharmacol. 2008, 61, 933–941. [Google Scholar] [CrossRef]
- Perumal, S.S.; Shanthi, P.; Sachdanandam, P. Augmented efficacy of tamoxifen in rat breast tumorigenesis when gavaged along with ribofavin, niacin, and CoQ10: Efects on lipid peroxidation and antioxidants in mitochondria. Chem. Biol. Interact. 2005, 152, 49–58. [Google Scholar] [CrossRef]
- Guthrie, N.; Gapor, A.; Chambers, A.F.; Carroll, K.K. Palm oil tocotrienols and plant flavonoids act synergistically with each other and with Tamoxifen in inhibiting proliferation and growth of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells in culture. Asia Pac. J. Clin. Nutr. 1997, 6, 41–45. [Google Scholar]
- Lawenda, B.D.; Kelly, K.M.; Ladas, E.J.; Sagar, S.M.; Vickers, A.; Blumberg, J.B. Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy. J. Natl. Cancer Inst. 2008, 100, 773–783. [Google Scholar] [CrossRef]
- Diao, Q.X.; Zhang, J.Z.; Zhao, T.; Xue, F.; Gao, F.; Ma, S.M.; Wang, Y. Vitamin E promotes breast cancer cell proliferation by reducing ROS production and p53 expression. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2710–2717. [Google Scholar]
- Chamras, H.; Barsky, S.H.; Ardashian, A.; Navasartian, D.; Heber, D.; Glaspy, J.A. Novel interactions of vitamin E and estrogen in breast cancer. Nutr. Cancer 2005, 52, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Peralta, E.A.; Viegas, M.L.; Louis, S.; Engle, D.L.; Dunnington, G.L. Effect of vitamin E on tamoxifen-treated breast cancer cells. Surgery 2006, 140, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Peralta, E.A.; Brewer, A.T.; Louis, S.; Dunnington, G.L. Vitamin E increases biomarkers of estrogen stimulation when taken with tamoxifen. J. Surg. Res. 2009, 153, 143–147. [Google Scholar] [CrossRef]
- Tiwary, R.; Yu, W.; de Graffenried, L.A.; Sanders, B.G.; Kline, K. Targeting cholesterol-rich microdomains to circumvent tamoxifen-resistant breast cancer. Breast Cancer Res. 2011, 13, R120. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.H.; Yang, S.Y.; You, S.L.; Lien, H.C.; Lin, C.H.; Lin, P.H.; Huang, C.S. Polymorphisms of ESR1, UGT1A1, HCN1, MAP3K1 and CYP2B6 are associated with the prognosis of hormone receptor positive early breast cancer. Oncotarget 2017, 8, 20925–20938. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khallouki, F.; Hajji, L.; Saber, S.; Bouddine, T.; Edderkaoui, M.; Bourhia, M.; Mir, N.; Lim, A.; El Midaoui, A.; Giesy, J.P.; et al. An Update on Tamoxifen and the Chemo-Preventive Potential of Vitamin E in Breast Cancer Management. J. Pers. Med. 2023, 13, 754. https://doi.org/10.3390/jpm13050754
Khallouki F, Hajji L, Saber S, Bouddine T, Edderkaoui M, Bourhia M, Mir N, Lim A, El Midaoui A, Giesy JP, et al. An Update on Tamoxifen and the Chemo-Preventive Potential of Vitamin E in Breast Cancer Management. Journal of Personalized Medicine. 2023; 13(5):754. https://doi.org/10.3390/jpm13050754
Chicago/Turabian StyleKhallouki, Farid, Lhoussain Hajji, Somayya Saber, Toufik Bouddine, Mouad Edderkaoui, Mohammed Bourhia, Nora Mir, Adrian Lim, Adil El Midaoui, John P. Giesy, and et al. 2023. "An Update on Tamoxifen and the Chemo-Preventive Potential of Vitamin E in Breast Cancer Management" Journal of Personalized Medicine 13, no. 5: 754. https://doi.org/10.3390/jpm13050754
APA StyleKhallouki, F., Hajji, L., Saber, S., Bouddine, T., Edderkaoui, M., Bourhia, M., Mir, N., Lim, A., El Midaoui, A., Giesy, J. P., Aboul-Soud, M. A. M., Silvente-Poirot, S., & Poirot, M. (2023). An Update on Tamoxifen and the Chemo-Preventive Potential of Vitamin E in Breast Cancer Management. Journal of Personalized Medicine, 13(5), 754. https://doi.org/10.3390/jpm13050754