

Prediction of Acute Cardiac Rejection Based on Gene Expression Profiles


Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection Criteria
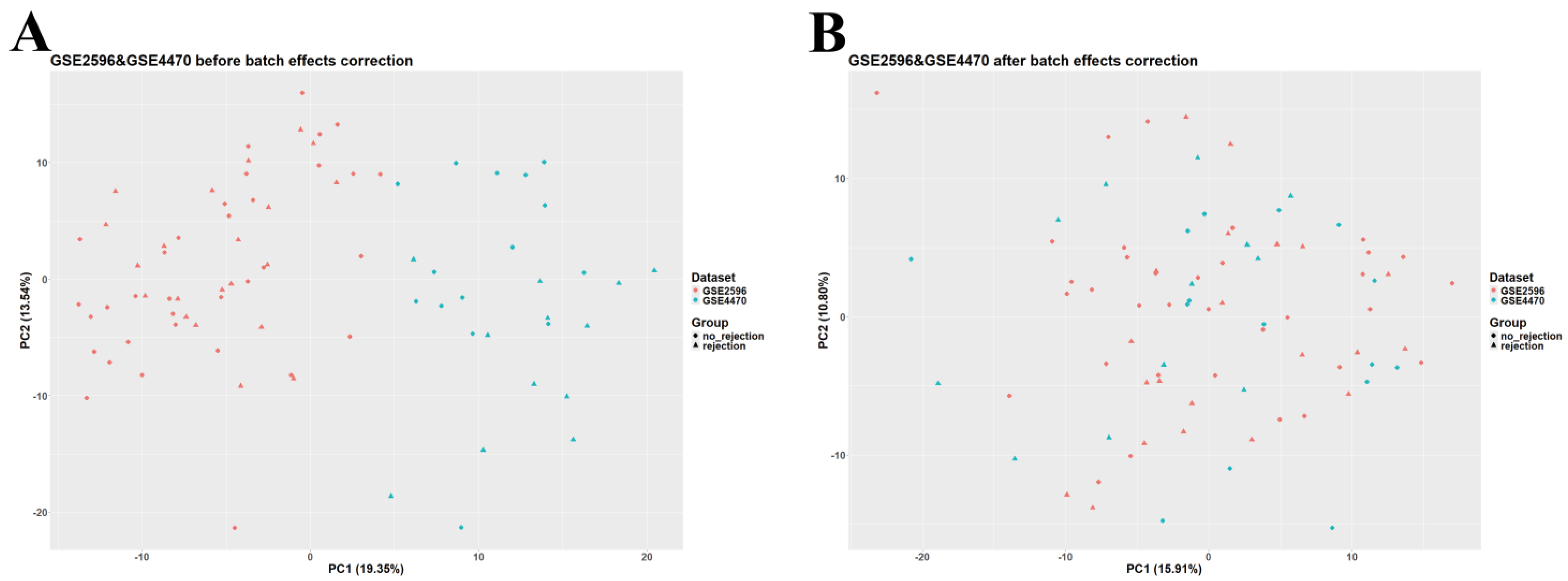
2.2. Data Preprocessing
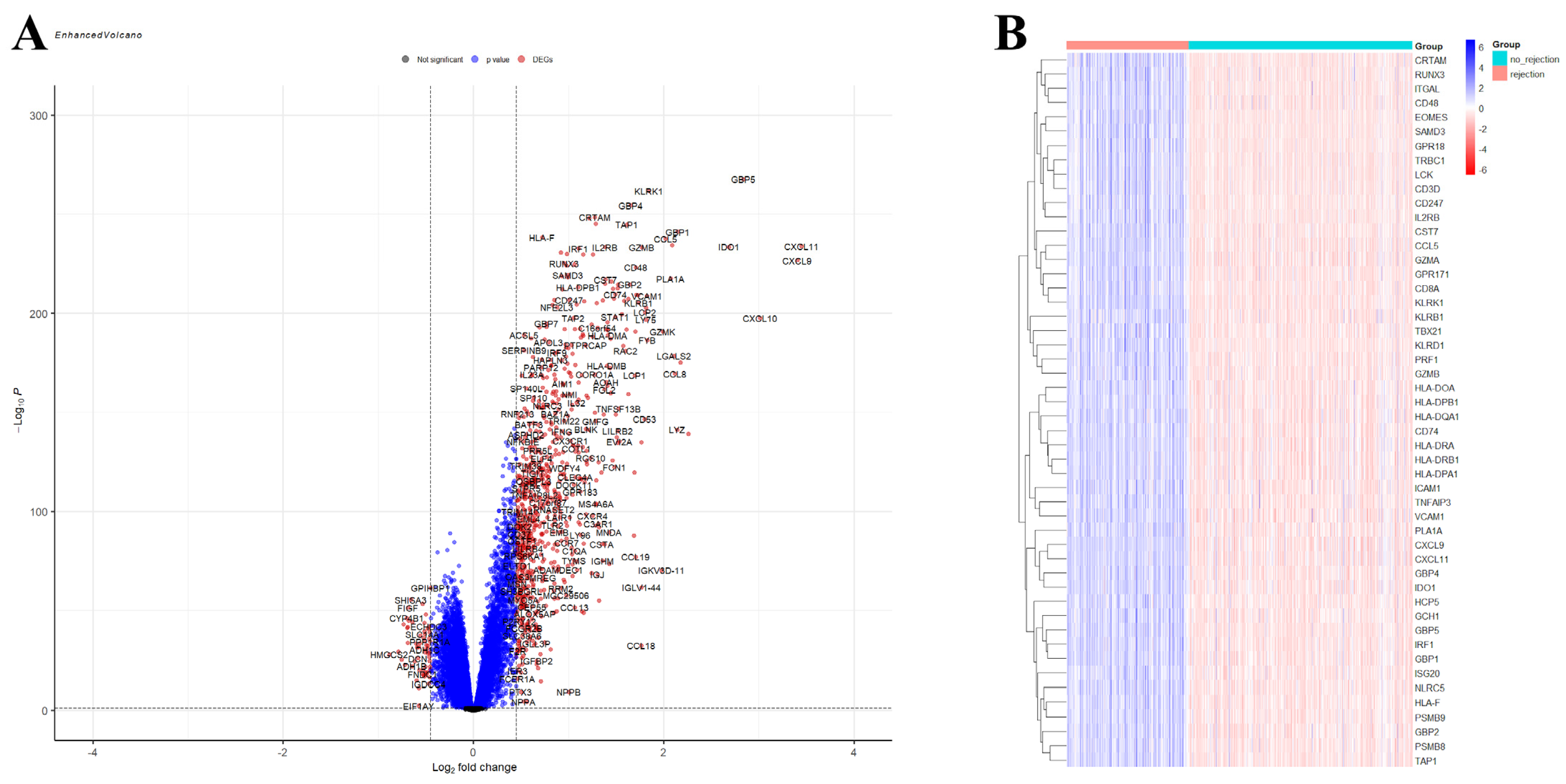
2.3. Identification of Differentially Expressed Genes
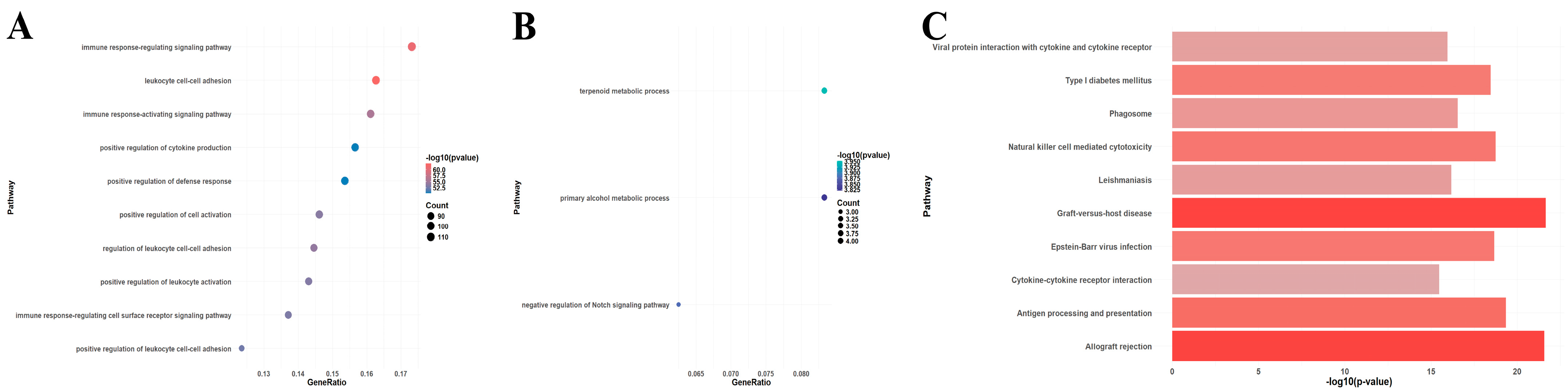
2.4. Enrichment Aanalysis
2.5. Data Preprocessing for Machine Learning Analysis
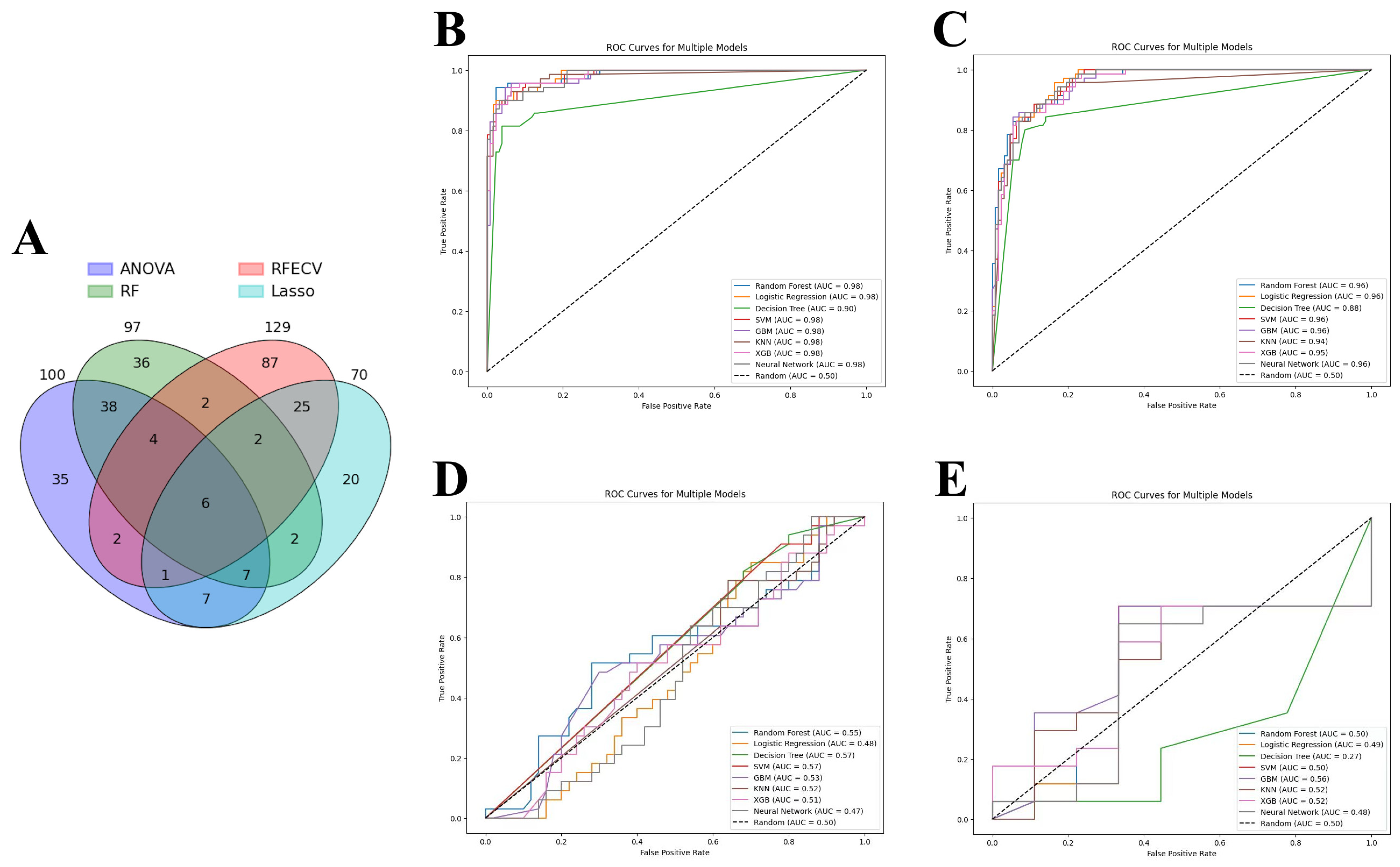
2.6. Feature Selection
- Analysis of variance (ANOVA) was leveraged to pinpoint the top 100 genes with significant expression differences between conditions, using SelectKBest with the f_classif score function. This approach narrows down the feature space to those most impactful for the analysis;
- Recursive feature elimination (RFE), through RFECV, combined with logistic regression and cross-validation (StratifiedKFold), dynamically identifies an optimal subset of features. Unlike traditional RFE which requires a predefined feature count, RFECV automatically determines the best number of features by maximizing cross-validation accuracy, making the selection process more data-driven;
- The least absolute shrinkage and selection operator (LASSO), applied via LassoCV, optimizes feature selection alongside model training by identifying non-zero coefficient features through cross-validation. This method effectively reduces the feature set to those most predictive of outcomes without pre-specifying a feature count;
- Random forest classifier (RFC) assesses feature importance after being trained with 50 trees. The optimal number of trees is found by using GridSearchCV. SelectFromModel with a ‘mean’ importance threshold is then used to filter the most significant features, allowing the model to concentrate on variables with the greatest impact on transplant outcomes.
2.7. Machine Learning Algorithms
2.8. Model Interpretation
3. Results
3.1. Identification of DEGs and Enrichment Analysis
3.2. Machine Learning Analysis
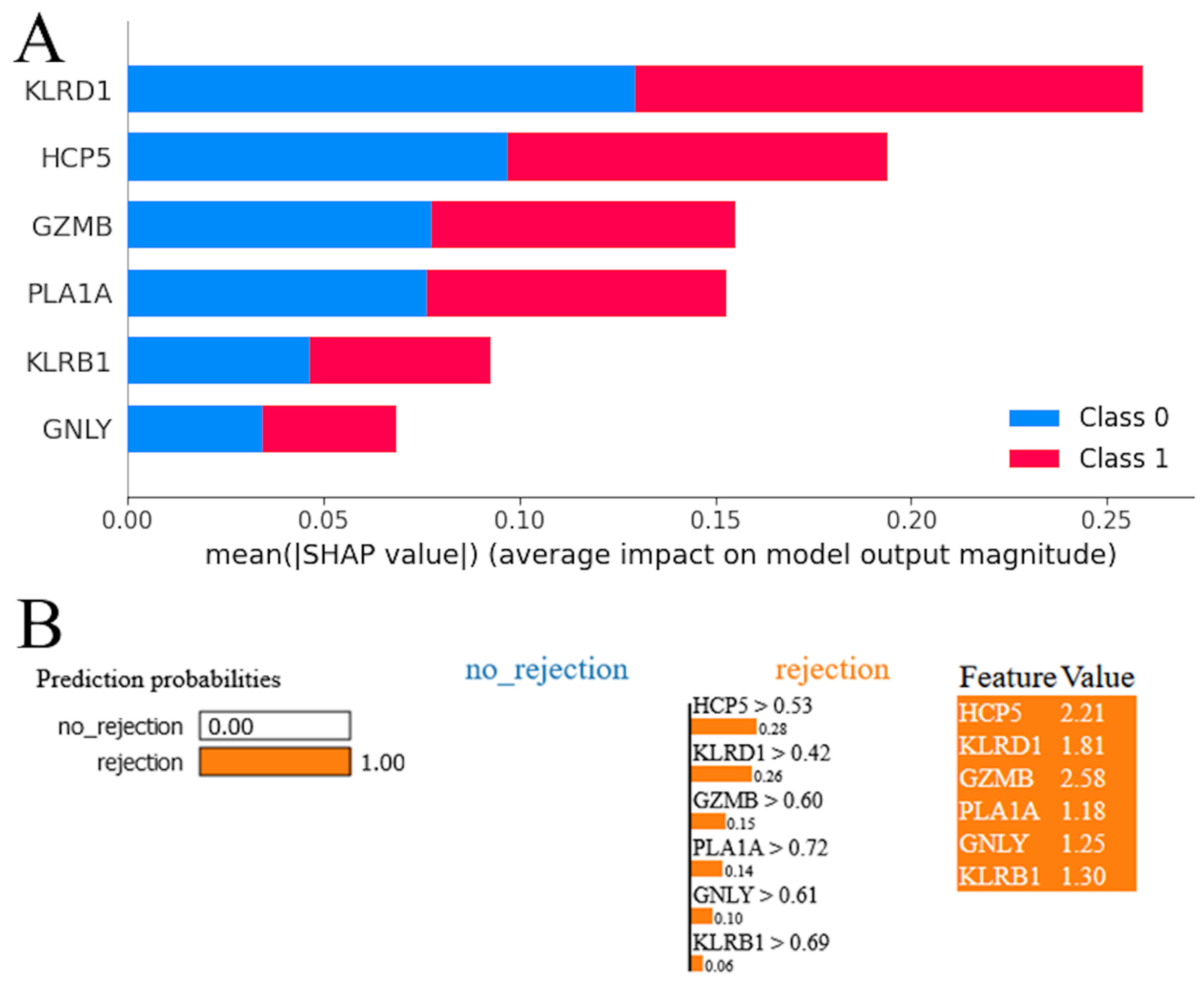
3.3. Model Interpretation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanveer, Y.; Arif, A.; Tsenteradze, T.; Anika, N.N.; Bakht, D.; Masood, Q.F.; Affaf, M.; Batool, W.; Yadav, I.; Gasim, R.W.; et al. Revolutionizing Heart Transplantation: A Multidisciplinary Approach to Xenotransplantation, Immunosuppression, Regenerative Medicine, Artificial Intelligence, and Economic Sustainability. Cureus 2023, 15, e46176. [Google Scholar] [CrossRef] [PubMed]
- Tonsho, M.; Michel, S.; Ahmed, Z.; Alessandrini, A.; Madsen, J.C. Heart transplantation: Challenges facing the field. Cold Spring Harb. Perspect. Med. 2014, 4, a015636. [Google Scholar] [CrossRef] [PubMed]
- Welch, T.S.; Mrsic, Z.; Mazimba, S. Monitoring for Rejection. In Contemporary Heart Transplantation; Bogar, L., Stempien-Otero, A., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–18. [Google Scholar]
- Hurskainen, M.; Ainasoja, O.; Lemström, K.B. Failing Heart Transplants and Rejection—A Cellular Perspective. J. Cardiovasc. Dev. Dis. 2021, 8, 180. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.C. Granzymes and perforin in solid organ transplant rejection. Cell Death Differ. 2010, 17, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Rascio, F.; Pontrelli, P.; Grandaliano, G. Cytotoxic T Lymphocytes (CTLs) and Kidney Transplantation: An Overview. In Cytotoxic T-Cells: Methods and Protocols; Gigante, M., Ranieri, E., Eds.; Springer: New York, NY, USA, 2021; pp. 203–213. [Google Scholar]
- Miyairi, S.; Baldwin, W.M.I.; Valujskikh, A.; Fairchild, R.L. Natural Killer Cells: Critical Effectors During Antibody-mediated Rejection of Solid Organ Allografts. Transplantation 2021, 105, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Faraco, J.; Lin, L.; Kornum, B.R.; Kenny, E.E.; Trynka, G.; Einen, M.; Rico, T.J.; Lichtner, P.; Dauvilliers, Y.; Arnulf, I.; et al. ImmunoChip Study Implicates Antigen Presentation to T Cells in Narcolepsy. PLOS Genet. 2013, 9, e1003270. [Google Scholar] [CrossRef] [PubMed]
- Albers, E.L.; Friedland-Little, J.M.; Hong, B.J.; Kemna, M.S.; Warner, P.; Law, Y.M. Human leukocyte antigen eplet mismatching is associated with increased risk of graft loss and rejection after pediatric heart transplant. Pediatr. Transplant. 2022, 26, e14126. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.T.; Tu, Z.; Alawieh, A.; Mulligan, J.; Esckilsen, S.; Quinn, K.; Sundararaj, K.; Wallace, C.; Finnegan, R.; Allen, P.; et al. Modulating donor mitochondrial fusion/fission delivers immunoprotective effects in cardiac transplantation. Am. J. Transplant. 2022, 22, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, L.A.; Bosch, L.; Kusters, P.J.H.; Lutgens, E.; Seijkens, T.T.P. The CD40-CD40L Dyad as Immunotherapeutic Target in Cardiovascular Disease. J. Cardiovasc. Transl. Res. 2021, 14, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Siemaszko, J.; Marzec-Przyszlak, A.; Bogunia-Kubik, K. NKG2D Natural Killer Cell Receptor—A Short Description and Potential Clinical Applications. Cells 2021, 10, 1420. [Google Scholar] [CrossRef]
- Halloran, P.F.; Famulski, K.S.; Reeve, J. Molecular assessment of disease states in kidney transplant biopsy samples. Nat. Rev. Nephrol. 2016, 12, 534–548. [Google Scholar] [CrossRef]
- Halloran, P.F.; Einecke, G.; Sikosana, M.L.N.; Madill-Thomsen, K. The Biology and Molecular Basis of Organ Transplant Rejection. In Pharmacology of Immunosuppression; Eisen, H.J., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–26. [Google Scholar]
- Shin, S.; Austin, P.C.; Ross, H.J.; Abdel-Qadir, H.; Freitas, C.; Tomlinson, G.; Chicco, D.; Mahendiran, M.; Lawler, P.R.; Billia, F.; et al. Machine learning vs. conventional statistical models for predicting heart failure readmission and mortality. ESC Heart Fail. 2021, 8, 106–115. [Google Scholar] [CrossRef]
- Singal, A.G.; Mukherjee, A.; Elmunzer, J.B.; Higgins, P.D.R.; Lok, A.S.; Zhu, J.; Marrero, J.A.; Waljee, A.K. Machine Learning Algorithms Outperform Conventional Regression Models in Predicting Development of Hepatocellular Carcinoma. Am. J. Gastroenterol. 2013, 108, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, M.; Vacchi, C.; Manfredi, A.; Cassone, G. Personalized Medicine and Machine Learning: A Roadmap for the Future. J. Clin. Med. 2022, 11, 4110. [Google Scholar] [CrossRef] [PubMed]
- Ozer, M.E.; Sarica, P.O.; Arga, K.Y. New Machine Learning Applications to Accelerate Personalized Medicine in Breast Cancer: Rise of the Support Vector Machines. OMICS 2020, 24, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.; Krzywinski, M.; Altman, N. Classification evaluation. Nat. Methods 2016, 13, 603–604. [Google Scholar] [CrossRef]
- Hossin, M.; Sulaiman, M.N. A Review On Evaluation Metrics For Data Classification Evaluations. Int. J. Min. Model. Manag. 2015, 5, 1–11. [Google Scholar]
- Chen, D.-P.; Chang, S.-W.; Jaing, T.-H.; Wang, W.-T.; Hsu, F.-P.; Tseng, C.-P. Single nucleotide polymorphisms within HLA region are associated with the outcomes of unrelated cord blood transplantation. Sci. Rep. 2021, 11, 21925. [Google Scholar] [CrossRef]
- Chen, D.-P.; Wen, Y.-H.; Wang, P.-N.; Hour, A.-L.; Lin, W.-T.; Hsu, F.-P.; Wang, W.-T. The adverse events of haematopoietic stem cell transplantation are associated with gene polymorphism within human leukocyte antigen region. Sci. Rep. 2021, 11, 1475. [Google Scholar] [CrossRef]
- Petersdorf, E.W.; Malkki, M.; Horowitz, M.M.; Spellman, S.R.; Haagenson, M.D.; Wang, T. Mapping MHC haplotype effects in unrelated donor hematopoietic cell transplantation. Blood 2013, 121, 1896–1905. [Google Scholar] [CrossRef]
- Duizendstra, A.A.; van der Grift, M.V.; Boor, P.P.; Noordam, L.; de Knegt, R.J.; Peppelenbosch, M.P.; Betjes, M.G.H.; Litjens, N.H.R.; Kwekkeboom, J. Current Tolerance-Associated Peripheral Blood Gene Expression Profiles After Liver Transplantation Are Influenced by Immunosuppressive Drugs and Prior Cytomegalovirus Infection. Front. Immunol. 2022, 12, 738837. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Ye, S.; Zhong, Z.; Zhou, X.; Zhou, W.; Liu, Z.; Lan, J.; Xiong, Y.; Ye, Q. Single-Cell Transcriptome Analysis of Chronic Antibody-Mediated Rejection After Renal Transplantation. Front. Immunol. 2022, 12, 767618. [Google Scholar] [CrossRef]
- Venner, J.M.; Hidalgo, L.G.; Famulski, K.S.; Chang, J.; Halloran, P.F. The Molecular Landscape of Antibody-Mediated Kidney Transplant Rejection: Evidence for NK Involvement Through CD16a Fc Receptors. Am. J. Transplant. 2015, 15, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Opelz, G.; Wiesel, M.; Ott, R.C.; Süsal, C. Serial Peripheral Blood Perforin and Granzyme B Gene Expression Measurements for Prediction of Acute Rejection in Kidney Graft Recipients. Am. J. Transplant. 2003, 3, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-L.; Wang, R.-l.; Liu, Z.; Wu, Q.; Li, X.-L.; He, Q.; Zhu, J.-Q. Granzyme B–Producing B Cells Function as a Feedback Loop for T Helper Cells in Liver Transplant Recipients with Acute Rejection. Inflammation 2021, 44, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Legros-Maïda, S.; Soulié, A.; Benvenuti, C.; Wargnier, A.; Vallée, N.; Berthou, C.; Guillet, J.; Sasportes, M.; Sigaux, N. Granzyme B and perforin can be used as predictive markers of acute rejection in heart transplantation. Eur. J. Immunol. 1994, 24, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Gao, P.; Liang, L.; Wang, Y.; Li, J.; Wang, J.; Hou, J.; Yang, C.; Wang, X. Noninvasive quantification of granzyme B in cardiac allograft rejection using targeted ultrasound imaging. Front. Immunol. 2023, 14, 1164183. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yi, L.; Wang, Y.; Wang, W.; Zhao, Q.; Song, Y.; Ding, M.; Deng, C.; Chen, Y.; Xie, Y.; et al. Granzyme B-responsive fluorescent probe for non-invasive early diagnosis of transplant rejection. Biosens. Bioelectron. 2023, 232, 115303. [Google Scholar] [CrossRef] [PubMed]
- Halloran, P.F.; Venner, J.M.; Madill-Thomsen, K.S.; Einecke, G.; Parkes, M.D.; Hidalgo, L.G.; Famulski, K.S. Review: The transcripts associated with organ allograft rejection. Am. J. Transplant. 2018, 18, 785–795. [Google Scholar] [CrossRef]
- Hò, G.T.; Celik, A.A.; Huyton, T.; Hiemisch, W.; Blasczyk, R.; Simper, G.S.; Bade-Doeding, C. NKG2A/CD94 Is a New Immune Receptor for HLA-G and Distinguishes Amino Acid Differences in the HLA-G Heavy Chain. Int. J. Mol. Sci. 2020, 21, 4362. [Google Scholar] [CrossRef]
- Lin, Z.; Bashirova, A.A.; Viard, M.; Garner, L.; Quastel, M.; Beiersdorfer, M.; Kasprzak, W.K.; Akdag, M.; Yuki, Y.; Ojeda, P.; et al. HLA class I signal peptide polymorphism determines the level of CD94/NKG2-HLA-E-mediated regulation of effector cell responses. Nat. Immunol. 2023, 24, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, H.; Maeda, A.; Lo, P.C.; Matsuura, R.; Esquivel, E.L.; Asada, M.; Sakai, R.; Nakahata, K.; Yamamichi, T.; Umeda, S.; et al. HLA-G1, but Not HLA-G3, Suppresses Human Monocyte/Macrophage-mediated Swine Endothelial Cell Lysis. Transplant. Proc. 2016, 48, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Kawamura, T.; Ueno, T.; Usui, N.; Eguchi, H.; Miyagawa, S. The suppression of inflammatory macrophage-mediated cytotoxicity and proinflammatory cytokine production by transgenic expression of HLA-E. Transpl. Immunol. 2013, 29, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Twito, T.; Joseph, J.; Mociornita, A.; Rao, V.; Ross, H.; Delgado, D.H. The 14-bp deletion in the HLA-G gene indicates a low risk for acute cellular rejection in heart transplant recipients. J. Heart Lung Transplant. 2011, 30, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Pabón, M.A.; Navarro, C.E.; Osorio, J.C.; Gómez, N.; Moreno, J.P.; Donado, A.F.; Pérez, H.C.; Lozano, E. Impact of human leukocyte antigen molecules E, F, and G on the outcome of transplantation. Transplant. Proc. 2014, 46, 2957–2965. [Google Scholar] [CrossRef] [PubMed]
- Lila, N.; Amrein, C.; Guillemain, R.; Chevalier, P.; Latremouille, C.; Fabiani, J.N.; Dausset, J.; Carosella, E.D.; Carpentier, A. Human leukocyte antigen-G expression after heart transplantation is associated with a reduced incidence of rejection. Circulation 2002, 105, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Calderon, F.; Kang, B.H.; Kyrysyuk, O.; Zheng, S.; Wang, H.; Mathewson, N.D.; Luoma, A.M.; Ning, X.; Pyrdol, J.; Cao, X.; et al. Targeting of the CD161 inhibitory receptor enhances T-cell-mediated immunity against hematological malignancies. Blood 2024, 143, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Wyrożemski, Ł.; Qiao, S.W. Immunobiology and conflicting roles of the human CD161 receptor in T cells. Scand. J. Immunol. 2021, 94, e13090. [Google Scholar] [CrossRef] [PubMed]
- Kurioka, A.; Cosgrove, C.; Simoni, Y.; van Wilgenburg, B.; Geremia, A.; Björkander, S.; Sverremark-Ekström, E.; Thurnheer, C.; Günthard, H.F.; Khanna, N.; et al. CD161 Defines a Functionally Distinct Subset of Pro-Inflammatory Natural Killer Cells. Front. Immunol. 2018, 9, 486. [Google Scholar] [CrossRef]
- Schmid, F.; Mayer, C.; Büttner-Herold, M.; von Hörsten, S.; Amann, K.; Daniel, C. CD161a-positive natural killer (NK) cells and α-smooth muscle actin-positive myofibroblasts were upregulated by extrarenal DPP4 in a rat model of acute renal rejection. Diabetes Res. Clin. Pract. 2021, 173, 108691. [Google Scholar] [CrossRef]
- Thieme, C.J.; Weist, B.J.D.; Mueskes, A.; Roch, T.; Stervbo, U.; Rosiewicz, K.; Wehler, P.; Stein, M.; Nickel, P.; Kurtz, A.; et al. The TreaT-Assay: A Novel Urine-Derived Donor Kidney Cell-Based Assay for Prediction of Kidney Transplantation Outcome. Sci. Rep. 2019, 9, 19037. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.K.; Allos, H.; Al Dulaijan, B.S.; Melhem, G.; Sulkaj, I.; Alhaddad, J.B.; Saad, A.J.; Deban, C.; Chu, P.; Choi, J.Y.; et al. mTORC1 Inhibition Protects Human Regulatory T Cells From Granzyme-B-Induced Apoptosis. Front. Immunol. 2022, 13, 899975. [Google Scholar] [CrossRef]
- Yadav, B.; Prasad, N.; Agrawal, V.; Agarwal, V.; Jain, M. Lower Circulating Cytotoxic T-Cell Frequency and Higher Intragraft Granzyme-B Expression Are Associated with Inflammatory Interstitial Fibrosis and Tubular Atrophy in Renal Allograft Recipients. Medicina 2023, 59, 1175. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Duong Van Huyen, J.P.; Hidalgo, L.; Reeve, J.; Racapé, M.; Aubert, O.; Venner, J.M.; Falmuski, K.; Bories, M.C.; Beuscart, T.; et al. Gene Expression Profiling for the Identification and Classification of Antibody-Mediated Heart Rejection. Circulation 2017, 135, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hasse, S.; Bourgoin, S.G. Phosphatidylserine-specific phospholipase A1: A friend or the devil in disguise. Prog. Lipid Res. 2021, 83, 101112. [Google Scholar] [CrossRef] [PubMed]
- Heldal, T.F.; Åsberg, A.; Ueland, T.; Reisaeter, A.V.; Pischke, S.E.; Mollnes, T.E.; Aukrust, P.; Hartmann, A.; Heldal, K.; Jenssen, T. Inflammation in the early phase after kidney transplantation is associated with increased long-term all-cause mortality. Am. J. Transplant. 2022, 22, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Kildey, K.; Francis, R.S.; Hultin, S.; Harfield, M.; Giuliani, K.; Law, B.M.P.; Wang, X.; See, E.J.; John, G.; Ungerer, J.; et al. Specialized Roles of Human Natural Killer Cell Subsets in Kidney Transplant Rejection. Front. Immunol. 2019, 10, 1877. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, K.; Ambrose, A.R.; Davis, D.M. Escaping Death: How Cancer Cells and Infected Cells Resist Cell-Mediated Cytotoxicity. Front. Immunol. 2022, 13, 867098. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lieberman, J. Knocking ‘em Dead: Pore-Forming Proteins in Immune Defense. Annu. Rev. Immunol. 2020, 38, 455–485. [Google Scholar] [CrossRef]
- Dotiwala, F.; Lieberman, J. Granulysin: Killer lymphocyte safeguard against microbes. Curr. Opin. Immunol. 2019, 60, 19–29. [Google Scholar] [CrossRef]
- Schilke, R.M.; Blackburn, C.M.R.; Bamgbose, T.T.; Woolard, M.D. Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis. Biomolecules 2020, 10, 1449. [Google Scholar] [CrossRef] [PubMed]
- van Smaalen, T.C.; Ellis, S.R.; Mascini, N.E.; Siegel, T.P.; Cillero-Pastor, B.; Hillen, L.M.; van Heurn, L.W.E.; Peutz-Kootstra, C.J.; Heeren, R.M.A. Rapid Identification of Ischemic Injury in Renal Tissue by Mass-Spectrometry Imaging. Anal. Chem. 2019, 91, 3575–3581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S.; Xu, Y.P.; Sui, X.L.; Zhang, Y.Z.; Gu, F.J.; Chen, J.H. Correlation between histone H3K4 trimethylation and DNA methylation and evaluation of the metabolomic features in acute rejection after kidney transplantation. Am. J. Transl. Res. 2020, 12, 7565–7580. [Google Scholar] [PubMed]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef]
- Smialowski, P.; Frishman, D.; Kramer, S. Pitfalls of supervised feature selection. Bioinformatics 2010, 26, 440–443. [Google Scholar] [CrossRef]
- AbdElNabi, M.L.R.; Wajeeh Jasim, M.; EL-Bakry, H.M.; Hamed, N.; Taha, M.; M. Khalifa, N.E. Breast and Colon Cancer Classification from Gene Expression Profiles Using Data Mining Techniques. Symmetry 2020, 12, 408. [Google Scholar] [CrossRef]
- Halloran, P.F.; Madill-Thomsen, K.; Aliabadi-Zuckermann, A.Z.; Cadeiras, M.; Crespo-Leiro, M.G.; Depasquale, E.C.; Deng, M.; Gökler, J.; Kim, D.H.; Kobashigawa, J.; et al. Many heart transplant biopsies currently diagnosed as no rejection have mild molecular antibody-mediated rejection-related changes. J. Heart Lung Transplant. 2022, 41, 334–344. [Google Scholar] [CrossRef]
- Halloran, P.F.; Potena, L.; Van Huyen, J.-P.D.; Bruneval, P.; Leone, O.; Kim, D.H.; Jouven, X.; Reeve, J.; Loupy, A. Building a tissue-based molecular diagnostic system in heart transplant rejection: The heart Molecular Microscope Diagnostic (MMDx) System. J. Heart Lung Transplant. 2017, 36, 1192–1200. [Google Scholar] [CrossRef]
- Suo, L.; Murillo, M.C.; Gallay, B.; Hod-Dvorai, R. Discrepancy Analysis between Histology and Molecular Diagnoses in Kidney Allograft Biopsies: A Single-Center Experience. Int. J. Mol. Sci. 2023, 24, 13817. [Google Scholar] [CrossRef]
- Randhawa, P. The MMDx® diagnostic system: A critical re-appraisal of its knowledge gaps and a call for rigorous validation studies. Clin. Transplant. 2022, 36, e14747. [Google Scholar] [CrossRef]
- Singh, D.; Singh, B. Investigating the impact of data normalization on classification performance. Appl. Soft Comput. 2020, 97, 105524. [Google Scholar] [CrossRef]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process Syst. 2017, 30, 4768–4777. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Number of Acute Cardiac Rejection Samples | Number of Non-Rejection Samples | Platform | Rejection Diagnosis | Set |
|---|---|---|---|---|---|
| GSE150059 | 853 | 467 | GPL16043 | MMDx | Training set, test set, internal validation set |
| GSE2596 | 35 | 21 | GPL1053 | Histology | External validation set 1 |
| GSE4470 | 15 | 12 | GPL1053 | Histology | External validation set 1 |
| GSE9377 | 17 | 9 | GPL887 | Histology | External validation set 2 |
| Metric | RF | LR | DT | SVM | GBM | KNN | XGB | MLP |
|---|---|---|---|---|---|---|---|---|
| Test set (MMDx) | ||||||||
| Accuracy | 0.95 | 0.95 | 0.91 | 0.93 | 0.92 | 0.93 | 0.94 | 0.93 |
| Precision | 0.95 | 0.95 | 0.92 | 0.93 | 0.92 | 0.93 | 0.95 | 0.90 |
| Recall | 0.90 | 0.90 | 0.81 | 0.89 | 0.86 | 0.89 | 0.89 | 0.90 |
| F1 Score | 0.93 | 0.93 | 0.86 | 0.91 | 0.89 | 0.91 | 0.92 | 0.90 |
| AUC | 0.98 | 0.98 | 0.90 | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 |
| MCC | 0.89 | 0.89 | 0.80 | 0.86 | 0.83 | 0.86 | 0.88 | 0.85 |
| AUPRC | 0.97 | 0.98 | 0.90 | 0.97 | 0.97 | 0.97 | 0.97 | 0.97 |
| Internal validation set (MMDx) | ||||||||
| Accuracy | 0.89 | 0.90 | 0.87 | 0.90 | 0.91 | 0.90 | 0.90 | 0.89 |
| Precision | 0.87 | 0.88 | 0.84 | 0.88 | 0.89 | 0.88 | 0.88 | 0.84 |
| Recall | 0.83 | 0.83 | 0.80 | 0.83 | 0.84 | 0.83 | 0.83 | 0.84 |
| F1 Score | 0.85 | 0.85 | 0.82 | 0.85 | 0.87 | 0.85 | 0.85 | 0.84 |
| AUC | 0.96 | 0.96 | 0.88 | 0.96 | 0.96 | 0.94 | 0.95 | 0.96 |
| MCC | 0.77 | 0.78 | 0.72 | 0.78 | 0.80 | 0.78 | 0.78 | 0.76 |
| AUPRC | 0.93 | 0.92 | 0.86 | 0.91 | 0.92 | 0.90 | 0.90 | 0.92 |
| Metric | RF | LR | DT | SVM | GBM | KNN | XGB | MLP |
|---|---|---|---|---|---|---|---|---|
| External validation set 1 (histology) | ||||||||
| Accuracy | 0.46 | 0.45 | 0.48 | 0.46 | 0.42 | 0.41 | 0.46 | 0.45 |
| Precision | 0.42 | 0.42 | 0.43 | 0.42 | 0.4 | 0.39 | 0.4 | 0.42 |
| Recall | 0.97 | 1 | 0.91 | 1 | 0.88 | 0.85 | 0.73 | 1 |
| F1 Score | 0.59 | 0.59 | 0.58 | 0.59 | 0.55 | 0.53 | 0.52 | 0.59 |
| AUC | 0.55 | 0.48 | 0.57 | 0.57 | 0.53 | 0.52 | 0.51 | 0.47 |
| MCC | 0.16 | 0.18 | 0.15 | 0.21 | 0 | −0.05 | 0.01 | 0.18 |
| AUPRC | 0.45 | 0.35 | 0.66 | 0.69 | 0.4 | 0.6 | 0.38 | 0.35 |
| External validation set 2 (histology) | ||||||||
| Accuracy | 0.65 | 0.54 | 0.27 | 0.35 | 0.69 | 0.54 | 0.42 | 0.54 |
| Precision | 0.75 | 0.73 | 0.33 | 0 | 0.8 | 0.73 | 0.62 | 0.73 |
| Recall | 0.71 | 0.47 | 0.12 | 0 | 0.71 | 0.47 | 0.29 | 0.47 |
| F1 Score | 0.73 | 0.57 | 0.17 | 0 | 0.75 | 0.57 | 0.4 | 0.57 |
| AUC | 0.5 | 0.49 | 0.27 | 0.5 | 0.56 | 0.52 | 0.52 | 0.48 |
| MCC | 0.26 | 0.13 | −0.37 | 0 | 0.36 | 0.13 | −0.04 | 0.13 |
| AUPRC | 0.67 | 0.66 | 0.53 | 0.66 | 0.7 | 0.66 | 0.72 | 0.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdrakhimov, B.; Kayewa, E.; Wang, Z. Prediction of Acute Cardiac Rejection Based on Gene Expression Profiles. J. Pers. Med. 2024, 14, 410. https://doi.org/10.3390/jpm14040410
Abdrakhimov B, Kayewa E, Wang Z. Prediction of Acute Cardiac Rejection Based on Gene Expression Profiles. Journal of Personalized Medicine. 2024; 14(4):410. https://doi.org/10.3390/jpm14040410
Chicago/Turabian StyleAbdrakhimov, Bulat, Emmanuel Kayewa, and Zhiwei Wang. 2024. "Prediction of Acute Cardiac Rejection Based on Gene Expression Profiles" Journal of Personalized Medicine 14, no. 4: 410. https://doi.org/10.3390/jpm14040410
APA StyleAbdrakhimov, B., Kayewa, E., & Wang, Z. (2024). Prediction of Acute Cardiac Rejection Based on Gene Expression Profiles. Journal of Personalized Medicine, 14(4), 410. https://doi.org/10.3390/jpm14040410