



Identifying Progression-Specific Alzheimer’s Subtypes Using Multimodal Transformer
 , ,
, ,  ,
,  , and
, and
Abstract
:1. Introduction
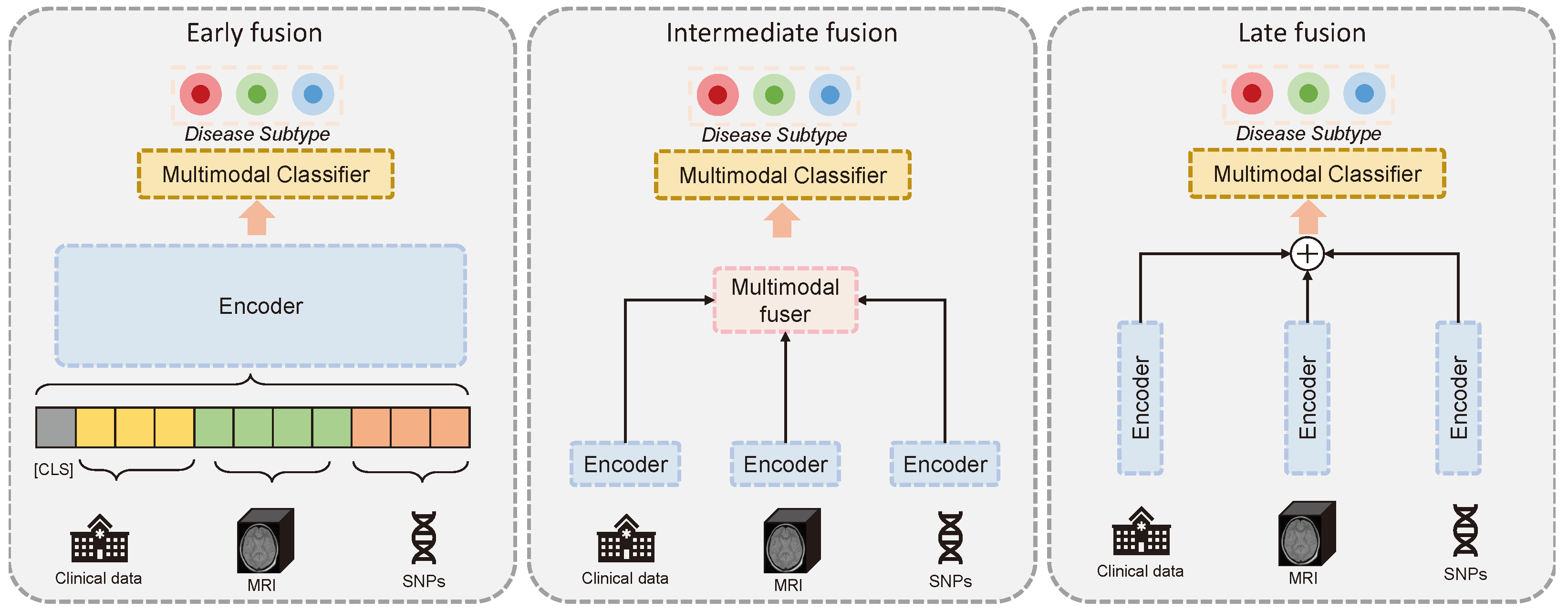
1.1. Multimodal Fusion
1.2. Overview and Contributions
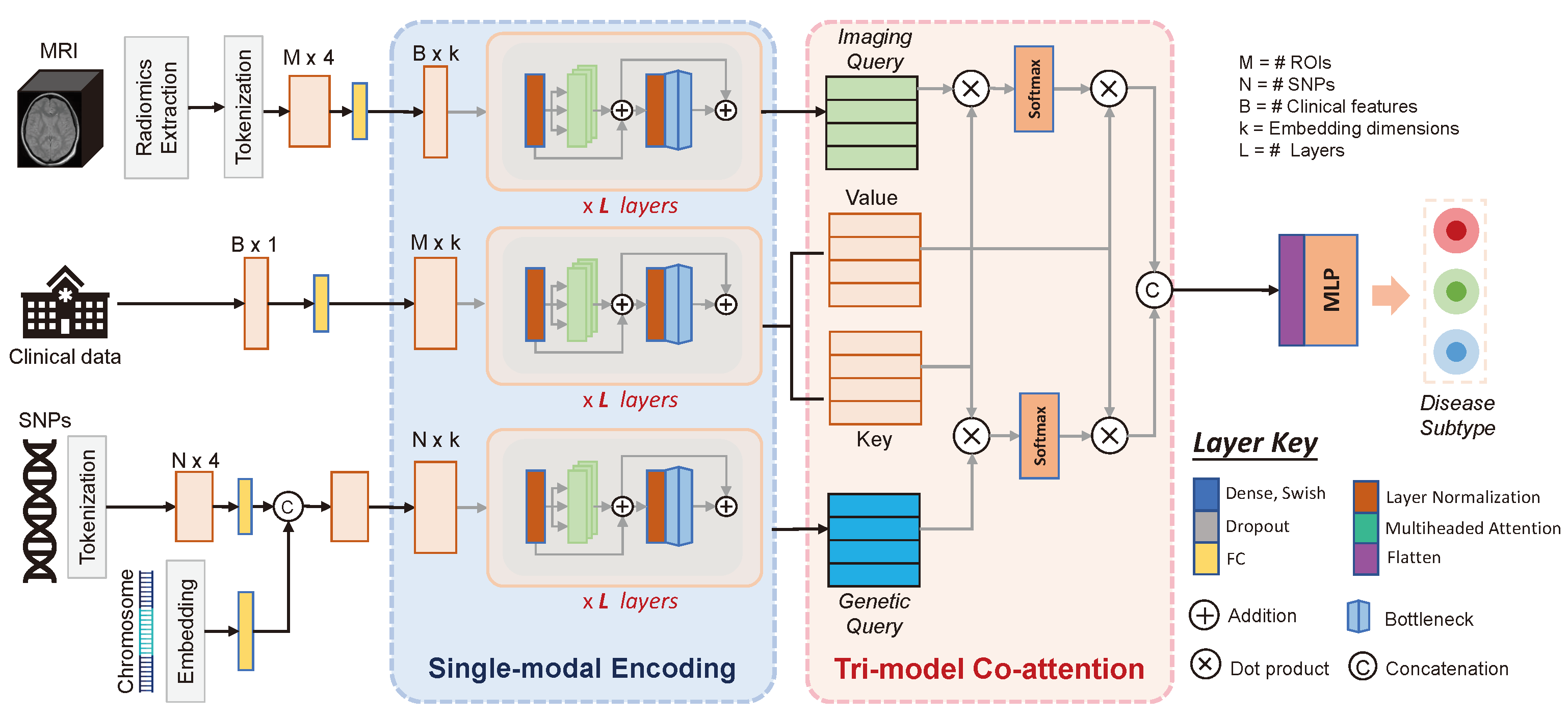
2. Method
2.1. Single-Modality Encoding
2.2. Tri-Modal Co-Attention
3. Materials and Experiments
3.1. Dataset
3.2. Experimental Design
4. Results and Discussion
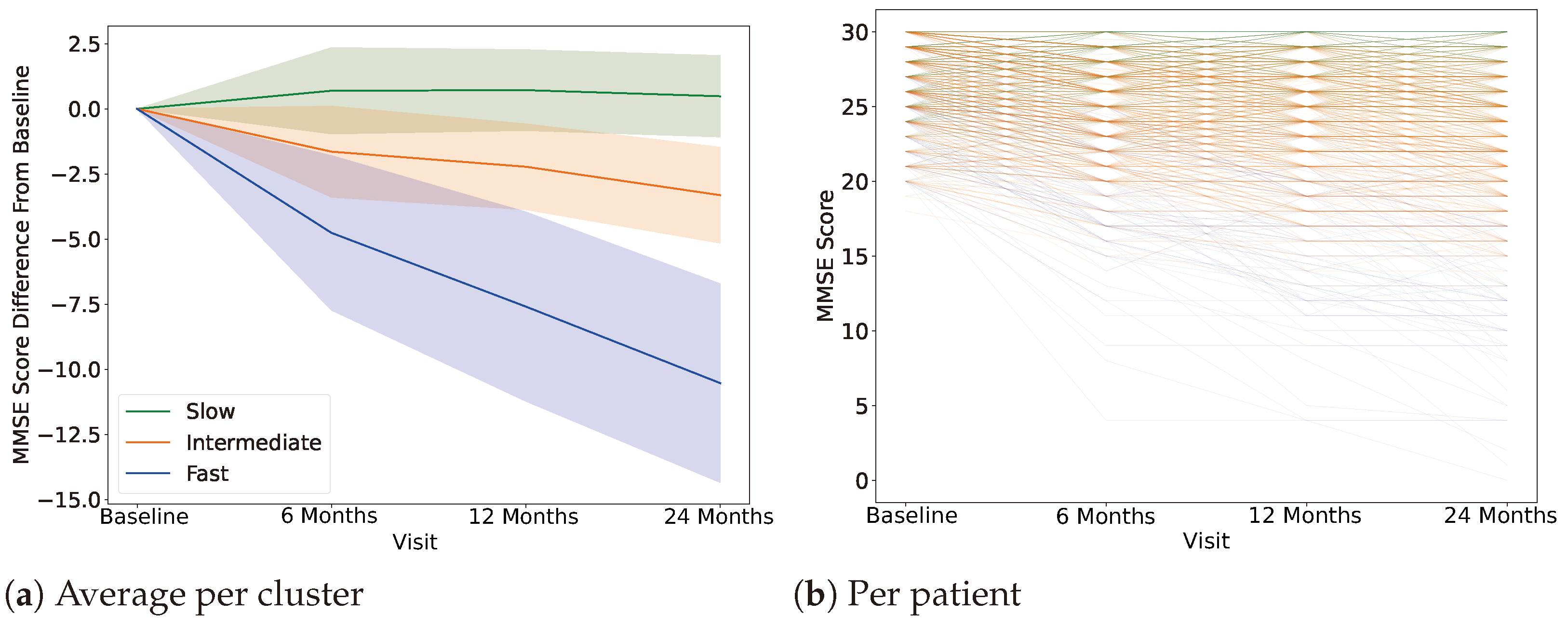
4.1. Clustering, Label Definition
4.2. AD Progression-Specific Subtype Classification
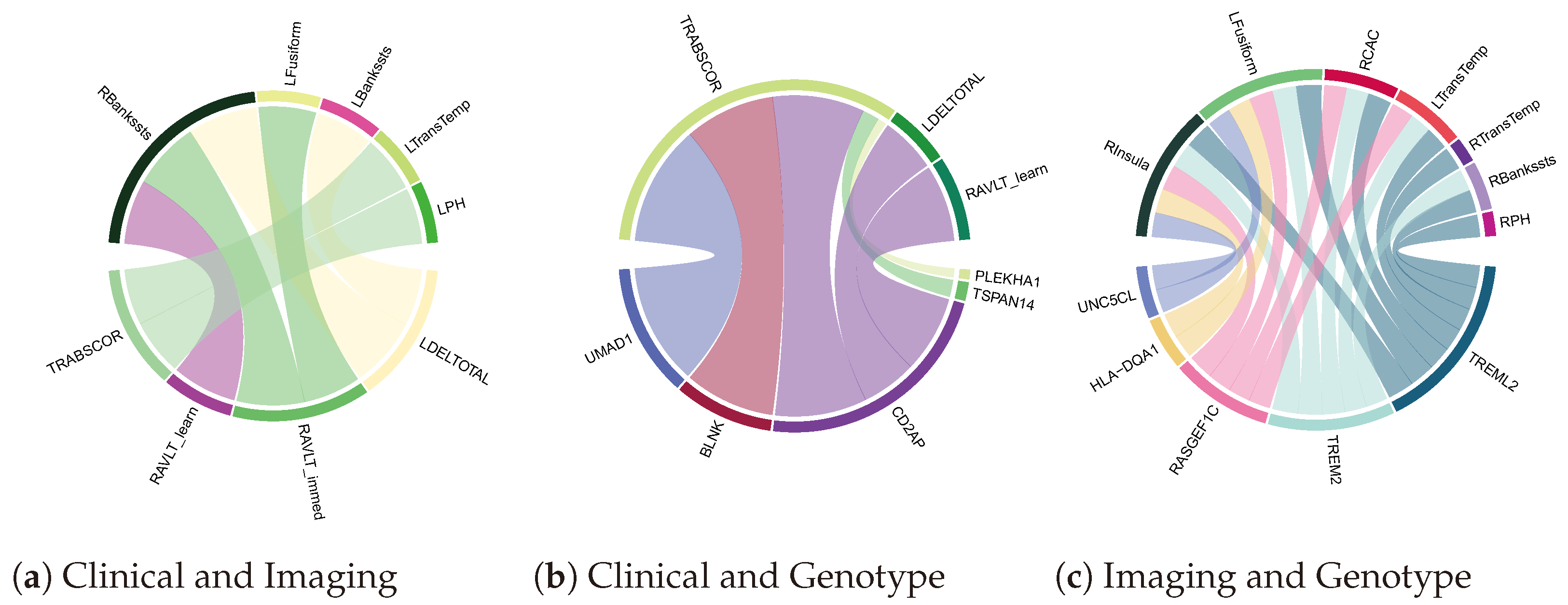
4.3. Biomarker Associations Learned by Co-Attention
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-AB protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Shcherbinin, S.; Evans, C.D.; Lu, M.; Andersen, S.W.; Pontecorvo, M.J.; Willis, B.A.; Gueorguieva, I.; Hauck, P.M.; Brooks, D.A.; Mintun, M.A.; et al. Association of Amyloid Reduction After Donanemab Treatment With Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Bondi, M.W.; Das, R.; Foss, M.; Hershey, L.A.; Koh, S.; Logan, R.; Poole, C.; Shega, J.W.; Sood, A.; et al. Quality improvement in neurology. Neurology 2019, 93, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Hassen, S.B.; Neji, M.; Hussain, Z.; Hussain, A.; Alimi, A.M.; Frikha, M. Deep learning methods for early detection of Alzheimer’s disease using structural MR images: A survey. Neurocomputing 2024, 576, 127325. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.Q.; Lin, R.R.; Wu, Z.Y. Early Diagnosis of Alzheimer’s Disease: Moving Toward a Blood-Based Biomarkers Era. Clin. Interv. Aging 2023, 18, 353–358. [Google Scholar] [CrossRef]
- Dubois, B.; von Arnim, C.A.F.; Burnie, N.; Bozeat, S.; Cummings, J. Biomarkers in Alzheimer’s disease: Role in early and differential diagnosis and recognition of atypical variants. Alzheimer’s Res. Ther. 2023, 15, 175. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, H.; Wang, W.; Wang, Z.; Luo, X.; Hramov, A.; Kurths, J. Edge-centric effective connection network based on muti-modal MRI for the diagnosis of Alzheimer’s disease. Neurocomputing 2023, 552, 126512. [Google Scholar] [CrossRef]
- Collij, L.E.; Salvadó, G.; Wottschel, V.; Mastenbroek, S.E.; Schoenmakers, P.; Heeman, F.; Aksman, L.; Wink, A.M.; Berckel, B.N.; van de Flier, W.M.; et al. Spatial-Temporal Patterns of B-Amyloid Accumulation. Neurology 2022, 98, e1692–e1703. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Mitelpunkt, A.; Galili, T.; Kozlovski, T.; Bregman, N.; Shachar, N.; Markus-Kalish, M.; Benjamini, Y. Novel Alzheimer’s disease subtypes identified using a data and knowledge driven strategy. Sci. Rep. 2020, 10, 1327. [Google Scholar] [CrossRef]
- Badhwar, A.; McFall, G.P.; Sapkota, S.; Black, S.E.; Chertkow, H.; Duchesne, S.; Masellis, M.; Li, L.; Dixon, R.A.; Bellec, P. A multiomics approach to heterogeneity in Alzheimer’s disease: Focused review and roadmap. Brain 2020, 143, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Martí-Juan, G.; Sanroma, G.; Piella, G.; Alzheimer’s Disease Neuroimaging Initiative and the Alzheimer’s Disease Metabolomics Consortium. Revealing heterogeneity of brain imaging phenotypes in Alzheimer’s disease based on unsupervised clustering of blood marker profiles. PLoS ONE 2019, 14, e0211121. [Google Scholar] [CrossRef]
- Feng, Y.; Kim, M.; Yao, X.; Liu, K.; Long, Q.; Shen, L.; for the Alzheimer’s Disease Neuroimaging Initiative. Deep multiview learning to identify imaging-driven subtypes in mild cognitive impairment. BMC Bioinform. 2022, 23, 402. [Google Scholar] [CrossRef]
- El-Sappagh, S.; Ali, F.; Abuhmed, T.; Singh, J.; Alonso, J.M. Automatic detection of Alzheimer’s disease progression: An efficient information fusion approach with heterogeneous ensemble classifiers. Neurocomputing 2022, 512, 203–224. [Google Scholar] [CrossRef]
- Emon, M.A.; Heinson, A.; Wu, P.; Domingo-Fernández, D.; Sood, M.; Vrooman, H.; Corvol, J.C.; Scordis, P.; Hofmann-Apitius, M.; Fröhlich, H. Clustering of Alzheimer’s and Parkinson’s disease based on genetic burden of shared molecular mechanisms. Sci. Rep. 2020, 10, 19097. [Google Scholar] [CrossRef]
- Wen, J.; Varol, E.; Sotiras, A.; Yang, Z.; Chand, G.B.; Erus, G.; Shou, H.; Abdulkadir, A.; Hwang, G.; Dwyer, D.B.; et al. Multi-scale semi-supervised clustering of brain images: Deriving disease subtypes. Med. Image Anal. 2022, 75, 102304. [Google Scholar] [CrossRef]
- Poulakis, K.; Pereira, J.B.; Muehlboeck, J.S.; Wahlund, L.O.; Smedby, O.; Volpe, G.; Masters, C.L.; Ames, D.; Niimi, Y.; Iwatsubo, T.; et al. Multi-cohort and longitudinal Bayesian clustering study of stage and subtype in Alzheimer’s disease. Nat. Commun. 2022, 13, 4566. [Google Scholar] [CrossRef] [PubMed]
- Odusami, M.; Maskeliūnas, R.; Damaševičius, R. Optimized Convolutional Fusion for Multimodal Neuroimaging in Alzheimer’s Disease Diagnosis: Enhancing Data Integration and Feature Extraction. J. Pers. Med. 2023, 13, 1496. [Google Scholar] [CrossRef]
- Dadu, A.; Satone, V.; Kaur, R.; Hashemi, S.H.; Leonard, H.; Iwaki, H.; Makarious, M.B.; Billingsley, K.J.; Bandres-Ciga, S.; Sargent, L.J.; et al. Identification and prediction of Parkinson’s disease subtypes and progression using machine learning in two cohorts. NPJ Parkinson’s Dis. 2022, 8, 1–12. [Google Scholar] [CrossRef]
- Su, C.; Hou, Y.; Xu, J.; Xu, J.; Brendel, M.; Maasch, J.R.M.A.; Bai, Z.; Zhang, H.; Zhu, Y.; Henchcliffe, C.; et al. Parkinson’s Disease Progression, 2022. Pages: 2021.07.18.21260731. medRxiv 2022, 2021.07.18.21260731. [Google Scholar] [CrossRef]
- Nguyen, N.D.; Wang, D. Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 2020, 16, e1007677. [Google Scholar] [CrossRef]
- Huang, S.C.; Pareek, A.; Seyyedi, S.; Banerjee, I.; Lungren, M.P. Fusion of medical imaging and electronic health records using deep learning: A systematic review and implementation guidelines. NPJ Digit. Med. 2020, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Alayrac, J.B.; Donahue, J.; Luc, P.; Miech, A.; Barr, I.; Hasson, Y.; Lenc, K.; Mensch, A.; Millican, K.; Reynolds, M.; et al. Flamingo: A Visual Language Model for Few-Shot Learning. arXiv 2022, arXiv:2204.14198. [Google Scholar] [CrossRef]
- Akbari, H.; Yuan, L.; Qian, R.; Chuang, W.H.; Chang, S.F.; Cui, Y.; Gong, B. VATT: Transformers for Multimodal Self-Supervised Learning from Raw Video, Audio and Text. arXiv 2021, arXiv:2104.11178. [Google Scholar] [CrossRef]
- Chen, R.J.; Lu, M.Y.; Weng, W.H.; Chen, T.Y.; Williamson, D.F.K.; Manz, T.; Shady, M.; Mahmood, F. Multimodal Co-Attention Transformer for Survival Prediction in Gigapixel Whole Slide Images. In Proceedings of the IEEE/CVF International Conference on Computer Vision (ICCV), Montreal, BC, Canada, 11–17 October 2021; pp. 4015–4025. [Google Scholar]
- Jack, C.R., Jr.; Bernstein, M.A.; Fox, N.C.; Thompson, P.; Alexander, G.; Harvey, D.; Borowski, B.; Britson, P.J.; Whitwell, J.L.; Ward, C.; et al. The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J. Magn. Reson. Imaging 2008, 27, 685–691. [Google Scholar] [CrossRef]
- Machado-Reyes, D.; Kim, M.; Chao, H.; Shen, L.; Yan, P. Connectome transformer with anatomically inspired attention for Parkinson’s diagnosis. In Proceedings of the 13th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, New York, NY, USA, 7–10 August 2022; BCB ’22. pp. 1–4. [Google Scholar] [CrossRef]
- Machado-Reyes, D.; Kim, M.; Chaoh, H.; Hahn, J.; Shen, L.; Yan, P. Genomics transformer for diagnosing Parkinson’s disease. In Proceedings of the 2022 IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), Ioannina, Greece, 27–30 September 2022; pp. 1–4, ISSN 2641-3604. [Google Scholar] [CrossRef]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer Normalization. arXiv 2016, arXiv:1607.06450. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is All you Need. In Proceedings of the Advances in Neural Information Processing Systems; Curran Associates, Inc.: Red Hook, NY, USA, 2017; Volume 30. [Google Scholar]
- Fischl, B. FreeSurfer. NeuroImage 2012, 62, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Fischl, B.; Dale, A.M. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. USA 2000, 97, 11050–11055. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.; Truran-Sacrey, D.; Raptentsetsang, S.; Simonson, A.; Mezher, A.; Schuff, N.; Weiner, M. UCSF Freesurfer Methods; ADNI Alzheimers Disease Neuroimaging Initiative: San Francisco, CA, USA, 2014. [Google Scholar]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Hand, D.J.; Till, R.J. A Simple Generalisation of the Area Under the ROC Curve for Multiple Class Classification Problems. Mach. Learn. 2001, 45, 171–186. [Google Scholar] [CrossRef]
- Zhou, T.; Thung, K.H.; Zhu, X.; Shen, D. Effective feature learning and fusion of multimodality data using stage-wise deep neural network for dementia diagnosis. Hum. Brain Mapp. 2019, 40, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Massman, P.; Dunn, J.K. A Method for Estimating Progression Rates in Alzheimer Disease. Arch. Neurol. 2001, 58, 449–454. [Google Scholar] [CrossRef]
- Doody, R.S.; Pavlik, V.; Massman, P.; Rountree, S.; Darby, E.; Chan, W. Predicting progression of Alzheimer’s disease. Alzheimer’s Res. Ther. 2010, 2, 2. [Google Scholar] [CrossRef]
- Prosser, A.; Evenden, D.; Holmes, R.; Kipps, C. Progression modelling of cognitive decline and associated FDG-PET imaging features in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, e045900. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Terada, S.; Sato, S.; Nagao, S.; Ikeda, C.; Shindo, A.; Hayashi, S.; Oshima, E.; Yokota, O.; Uchitomi, Y. Trail Making Test B and brain perfusion imaging in mild cognitive impairment and mild Alzheimer’s disease. Psychiatry Res. Neuroimaging 2013, 213, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Matías-Guiu, J.A.; Cabrera-Martín, M.N.; Valles-Salgado, M.; Pérez-Pérez, A.; Rognoni, T.; Moreno-Ramos, T.; Carreras, J.L.; Matías-Guiu, J. Neural Basis of Cognitive Assessment in Alzheimer Disease, Amnestic Mild Cognitive Impairment, and Subjective Memory Complaints. Am. J. Geriatr. Psychiatry 2017, 25, 730–740. [Google Scholar] [CrossRef]
- Camacho, J.; Rábano, A.; Marazuela, P.; Bonaterra-Pastra, A.; Serna, G.; Moliné, T.; Ramón y Cajal, S.; Martínez-Sáez, E.; Hernández-Guillamon, M. Association of CD2AP neuronal deposits with Braak neurofibrillary stage in Alzheimer’s disease. Brain Pathol. 2021, 32, e13016. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slow | Intermediate | Fast | |
|---|---|---|---|
| Participants | 177 | 302 | 15 |
| MMSE (Baseline) | 27.35 ± 2.51 | 27.66 ± 1.86 | 24.93 ± 3.55 |
| MMSE (24 months) | 28.15± 2.15 | 23.86 ± 3.68 | 15.9 ± 4.84 |
| Age | 73.26 ± 7.82 | 72.44 ± 7.55 | 71.22 ± 3.922 |
| Sex | M: 102 F: 75 | M: 185 F: 117 | M: 9 F: 6 |
| Method | Full | Imaging | Genetics | Clinical |
|---|---|---|---|---|
| SVM | 0.705 ± 0.036 | 0.669 ± 0.060 | 0.525 ± 0.034 | 0.639 ± 0.078 |
| RF | 0.684 ± 0.048 | 0.677 ± 0.052 | 0.505 ± 0.031 | 0.659 ± 0.087 |
| Stage-wise fusion | 0.641 ± 0.017 | 0.557 ± 0.096 | 0.562 ± 0.078 | 0.655 ± 0.057 |
| Tri-COAT | 0.734 ± 0.076 | 0.648 ± 0.056 | 0.539 ± 0.084 | 0.697 ± 0.063 |
| Method | AUROC |
|---|---|
| Early | 0.571 ± 0.053 |
| Late | 0.604 ± 0.048 |
| Tri-COAT | 0.734 ± 0.076 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado Reyes, D.; Chao, H.; Hahn, J.; Shen, L.; Yan, P.; for the Alzheimer’s Disease Neuroimaging Initiative. Identifying Progression-Specific Alzheimer’s Subtypes Using Multimodal Transformer. J. Pers. Med. 2024, 14, 421. https://doi.org/10.3390/jpm14040421
Machado Reyes D, Chao H, Hahn J, Shen L, Yan P, for the Alzheimer’s Disease Neuroimaging Initiative. Identifying Progression-Specific Alzheimer’s Subtypes Using Multimodal Transformer. Journal of Personalized Medicine. 2024; 14(4):421. https://doi.org/10.3390/jpm14040421
Chicago/Turabian StyleMachado Reyes, Diego, Hanqing Chao, Juergen Hahn, Li Shen, Pingkun Yan, and for the Alzheimer’s Disease Neuroimaging Initiative. 2024. "Identifying Progression-Specific Alzheimer’s Subtypes Using Multimodal Transformer" Journal of Personalized Medicine 14, no. 4: 421. https://doi.org/10.3390/jpm14040421
APA StyleMachado Reyes, D., Chao, H., Hahn, J., Shen, L., Yan, P., & for the Alzheimer’s Disease Neuroimaging Initiative. (2024). Identifying Progression-Specific Alzheimer’s Subtypes Using Multimodal Transformer. Journal of Personalized Medicine, 14(4), 421. https://doi.org/10.3390/jpm14040421