Interaction between Fexofenadine and CYP Phenotyping Probe Drugs in Geneva Cocktail

 ,
, 

Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Study Design
2.3. Analytical Methods
2.4. Genotyping
- (a)
- homozygous wild-type carriers 2677GG/3435CC (n = 4) and homozygous wild-type carriers at one chromosome and heterozygous for the other ex.2677GG/3435CT or 2677GT/3435CC (n = 6).
- (b)
- heterozygous carriers at both chromosomes (2677GT/3435CT) (n = 10) and homozygous mutated at one chromosome + homozygous wild-type for the other ex. 2677TT/3435CC or 2677GG/3435TT (n = 6)
- (c)
- homozygous mutated carriers at both chromosomes (2677TT/3435TT) (n = 3) and 2677GT/3435TT (n = 1)
2.5. Statistical Analysis
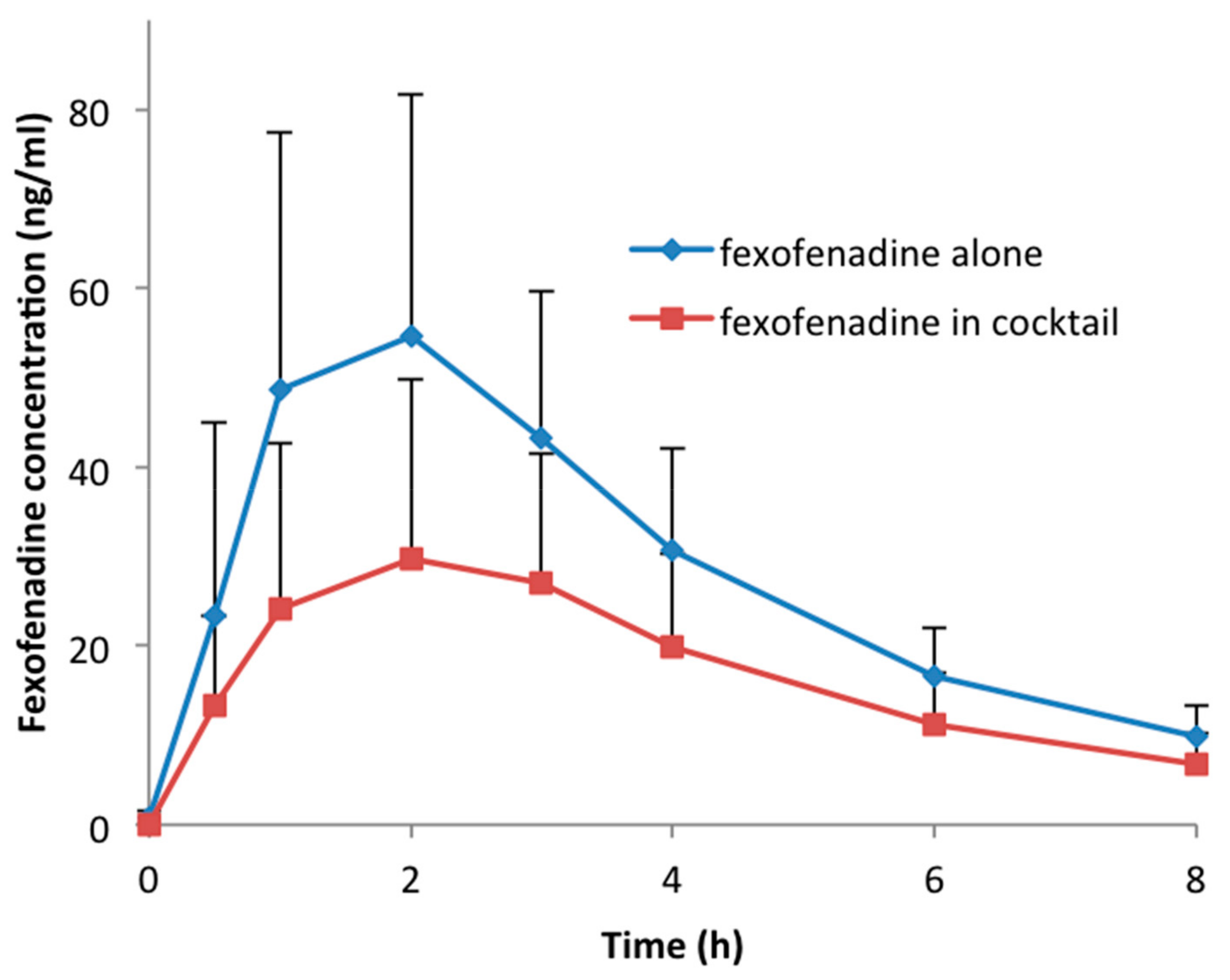
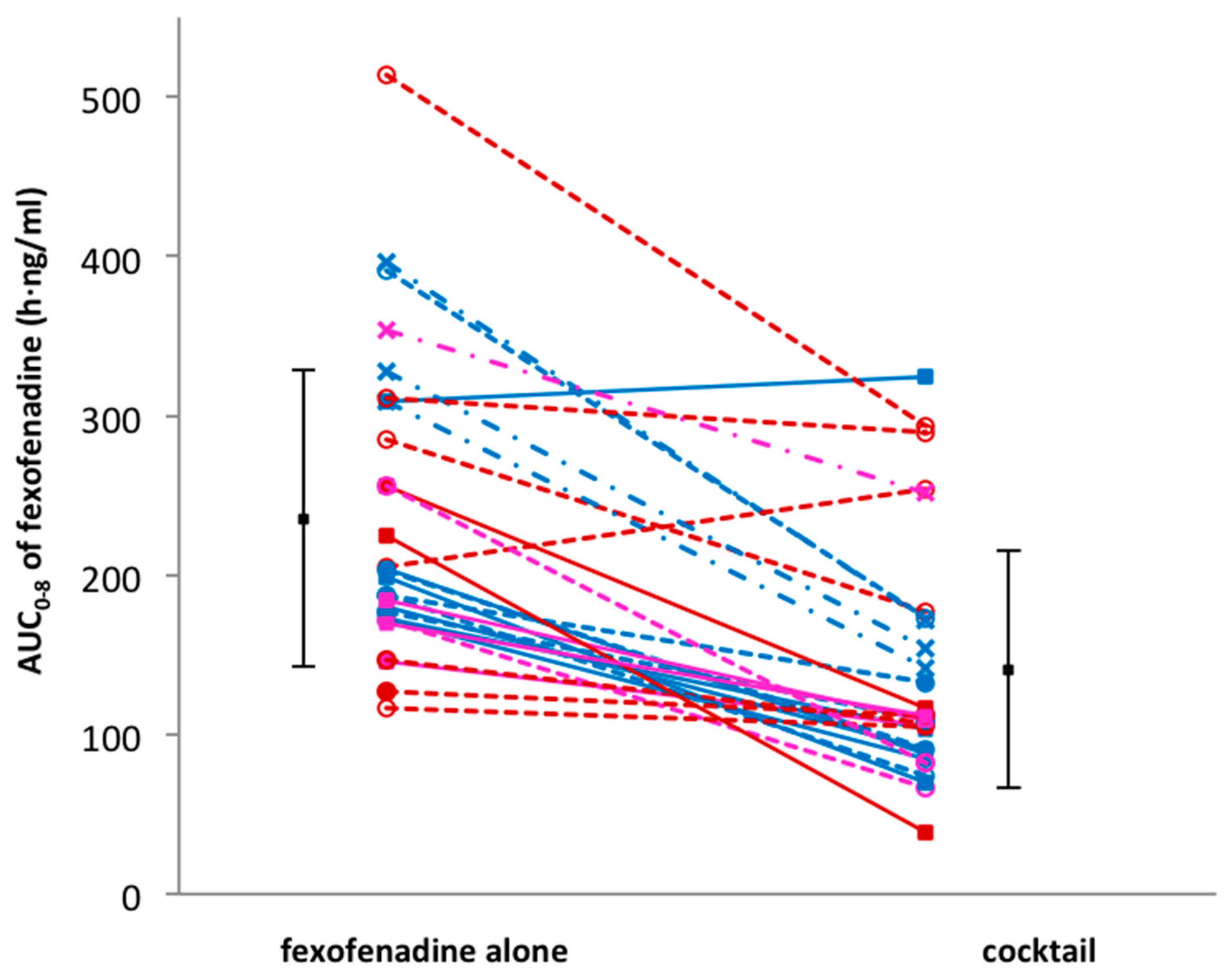
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Goh, B.C.; Reddy, N.J.; Dandamudi, U.B.; Laubscher, K.H.; Peckham, T.; Hodge, J.P.; Suttle, A.B.; Arumugham, T.; Xu, Y.; Xu, C.F.; et al. An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin. Pharmacol. Ther. 2010, 88, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Inui, N.; Akamatsu, T.; Uchida, S.; Tanaka, S.; Namiki, N.; Karayama, M.; Chida, K.; Watanabe, H. Chronological Effects of Rifampicin Discontinuation on Cytochrome P450 Activity in Healthy Japanese Volunteers, Using the Cocktail Method. Clin. Pharmacol. Ther. 2013, 94, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Shelepova, T.; Nafziger, A.N.; Victory, J.; Kashuba, A.D.; Rowland, E.; Zhang, Y.; Sellers, E.; Kearns, G.; Leeder, J.S.; Gaedigk, A.; et al. Effect of a triphasic oral contraceptive on drug-metabolizing enzyme activity as measured by the validated Cooperstown 5 + 1 cocktail. J. Clin. Pharmacol. 2005, 45, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Zadoyan, G.; Rokitta, D.; Klement, S.; Dienel, A.; Hoerr, R.; Gramatté, T.; Fuhr, U. Effect of Ginkgo biloba special extract EGb 761 (R) on human cytochrome P450 activity: A cocktail interaction study in healthy volunteers. Eur. J. Clin. Pharmacol. 2012, 68, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Muller, F.; Fromm, M.F. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, H. Importance of P-glycoprotein for drug-drug interactions. Handb. Exp. Pharmacol. 2011, 201, 285–297. [Google Scholar]
- EMEA. Guideline on the Investigation of Drug Interactions. 2012. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (accessed on 4 May 2016).
- FDA. Drug Interaction Studies-Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. 2012. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf (accessed on 4 May 2016).
- Ma, J.D.; Tsunoda, S.M.; Bertino, J.S.; Trivedi, M.; Beale, K.K.; Nafziger, A.N. Evaluation of in vivo P-glycoprotein phenotyping probes: A need for validation. Clin. Pharmacokinet. 2010, 49, 223–237. [Google Scholar] [CrossRef]
- Dumond, J.B.; Vourvahis, M.; Rezk, N.L.; Patterson, K.B.; Tien, H.C.; White, N.; Jennings, S.H.; Choi, S.O.; Li, J.; Wagner, M.J.; et al. A phenotype-genotype approach to predicting CYP450 and P-glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin. Pharmacol. Ther. 2010, 87, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Bosilkovska, M.; Samer, C.F.; Deglon, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Walder, B.; Desmeules, J.A.; Daali, Y. Geneva cocktail for cytochrome p450 and p-glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 2014, 96, 349–359. [Google Scholar] [CrossRef]
- Bosilkovska, M.; Déglon, J.; Samer, C.; Walder, B.; Desmeules, J.; Staub, C.; Daali, Y. Simultaenous LC-MS/MS quantification of P-glycoprotein and cytochrome P450 probe substrates and their metabolites in DBS and plasma. Bioanalysis 2014, 6, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Bosilkovska, M.; Samer, C.; Déglon, J.; Thomas, A.; Walder, B.; Desmeules, J.; Daali, Y. Evaluation of Mutual Drug-Drug Interaction within Geneva Cocktail for Cytochrome P450 Phenotyping using Innovative Dried Blood Sampling Method. Basic Clin. Pharmacol. 2016, 119, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joy, M.S.; Frye, R.F.; Nolin, T.D.; Roberts, B.V.; La, M.K.; Wang, J.; Brouwer, K.L.; Dooley, M.A.; Falk, R.J. In vivo alterations in drug metabolism and transport pathways in patients with chronic kidney diseases. Pharmacotherapy 2014, 34, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hamman, M.A.; Bruce, M.A.; Haehner-Daniels, B.D.; Hall, S.D. The effect of rifampin administration on the disposition of fexofenadine. Clin. Pharmacol. Ther. 2001, 69, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Yasui-Furukori, N.; Uno, T.; Sugawara, K.; Tateishi, T. Different effects of three transporting inhibitors, verapamil, cimetidine, and probenecid, on fexofenadine pharmacokinetics. Clin. Pharmacol. Ther. 2005, 77, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Park, P.W.; Park, J.Y. Short-term effect of quercetin on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein, in healthy volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Uno, T.; Sugawara, K.; Tateishi, T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br. J. Clin. Pharmacol. 2006, 61, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Akamine, Y.; Miura, M.; Yasui-Furukori, N.; Ieiri, I.; Uno, T. Effects of multiple-dose rifampicin 450 mg on the pharmacokinetics of fexofenadine enantiomers in Japanese volunteers. J. Clin. Pharm. Ther. 2015, 40, 98–103. [Google Scholar] [CrossRef]
- Matsushima, S.; Maeda, K.; Ishiguro, N.; Igarashi, T.; Sugiyama, Y. Investigation of the inhibitory effects of various drugs on the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2008, 36, 663–669. [Google Scholar] [CrossRef]
- Soldner, A.; Christians, U.; Susanto, M.; Wacher, V.J.; Silverman, J.A.; Benet, L.Z. Grapefruit juice activates P-glycoprotein-mediated drug transport. Pharm. Res. 1999, 16, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Kindla, J.; Muller, F.; Mieth, M.; Fromm, M.F.; Konig, J. Influence of non-steroidal anti-inflammatory drugs on organic anion transporting polypeptide (OATP) 1B1- and OATP1B3-mediated drug transport. Drug Metab. Dispos. 2011, 39, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.J.; Collier, A.C.; Kharasch, E.D.; Whittington, D.; Thummel, K.E.; Unadkat, J.D. Complex drug interactions of the HIV protease inhibitors 3: Effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab. Dispos. 2012, 40, 610–616. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yu, Y.; Prasad, B.; Chen, X.; Unadkat, J.D. Mechanism of an unusual, but clinically significant, digoxin-bupropion drug interaction. Biopharm. Drug Dispos. 2014, 35, 253–263. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genotype Group | G2677T/A | C3435T | Number of Subjects |
|---|---|---|---|
| (a) | GG | CC | 4 |
| GG | CT | 5 | |
| GT | CC | 1 | |
| (b) | GT | CT | 10 |
| TT | CC | 4 | |
| GG | TT | 2 | |
| (c) | GT | TT | 1 |
| TT | TT | 3 |
| Fexofenadine Alone | Fexofenadine in Cocktail | Geometric Mean Ratio | 90% CI | p-Value | |
|---|---|---|---|---|---|
| t1/2 (h) | 2.46 ± 0.30 | 2.64 ± 0.55 | 1.07 | 1.00–1.13 | 0.054 |
| Tmax (h) | 1.60 ± 0.66 | 2.01 ± 0.98 | 1.14 | 0.87–1.50 | 0.082 |
| Cmax (ng/mL) | 59.6 ± 27.0 | 32.9 ± 21.8 | 0.51 | 0.44–0.59 | <0.001 |
| AUC0–8 (h·ng/mL) | 235.5 ± 92.8 | 140.8 ± 74.2 | 0.57 | 0.50–0.65 | <0.001 |
| AUC0–∞ (h·ng/mL) | 270.4 ± 102.3 | 165.7 ± 84.4 | 0.59 | 0.52–0.67 | <0.001 |
| Cl/F (l/h) | 103.9 ± 33.7 | 189.1 ± 95.1 | 1.70 | 1.49–1.93 | <0.001 |
| Genotype Group | AUC0–8 Fexofenadine Alone | AUC0–8 Fexofenadine in Cocktail |
|---|---|---|
| a (n = 10) | 207.3 ± 44.9 | 113.7 ± 77.5 |
| b (n = 16) | 225.4 ± 105.9 | 147.9 ± 74.9 |
| c (n = 4) | 346.7 ± 37.8 | 179.9 ± 49.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosilkovska, M.; Magliocco, G.; Desmeules, J.; Samer, C.; Daali, Y. Interaction between Fexofenadine and CYP Phenotyping Probe Drugs in Geneva Cocktail. J. Pers. Med. 2019, 9, 45. https://doi.org/10.3390/jpm9040045
Bosilkovska M, Magliocco G, Desmeules J, Samer C, Daali Y. Interaction between Fexofenadine and CYP Phenotyping Probe Drugs in Geneva Cocktail. Journal of Personalized Medicine. 2019; 9(4):45. https://doi.org/10.3390/jpm9040045
Chicago/Turabian StyleBosilkovska, Marija, Gaelle Magliocco, Jules Desmeules, Caroline Samer, and Youssef Daali. 2019. "Interaction between Fexofenadine and CYP Phenotyping Probe Drugs in Geneva Cocktail" Journal of Personalized Medicine 9, no. 4: 45. https://doi.org/10.3390/jpm9040045
APA StyleBosilkovska, M., Magliocco, G., Desmeules, J., Samer, C., & Daali, Y. (2019). Interaction between Fexofenadine and CYP Phenotyping Probe Drugs in Geneva Cocktail. Journal of Personalized Medicine, 9(4), 45. https://doi.org/10.3390/jpm9040045