Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Microbial Growth Conditions
2.2. Immunization of Insects
2.3. Transcriptome Sequencing, De Novo Assembly and Functional Annotation
2.4. Gene Expression Analysis
2.5. AMP Screening and Prediction
2.6. Peptide Synthesis
2.7. Antimicrobial Activity Assay
2.8. Hemolytic Assay
2.9. Data Availability
3. Result and Discussion
3.1. Transcriptome Sequencing and Assembly
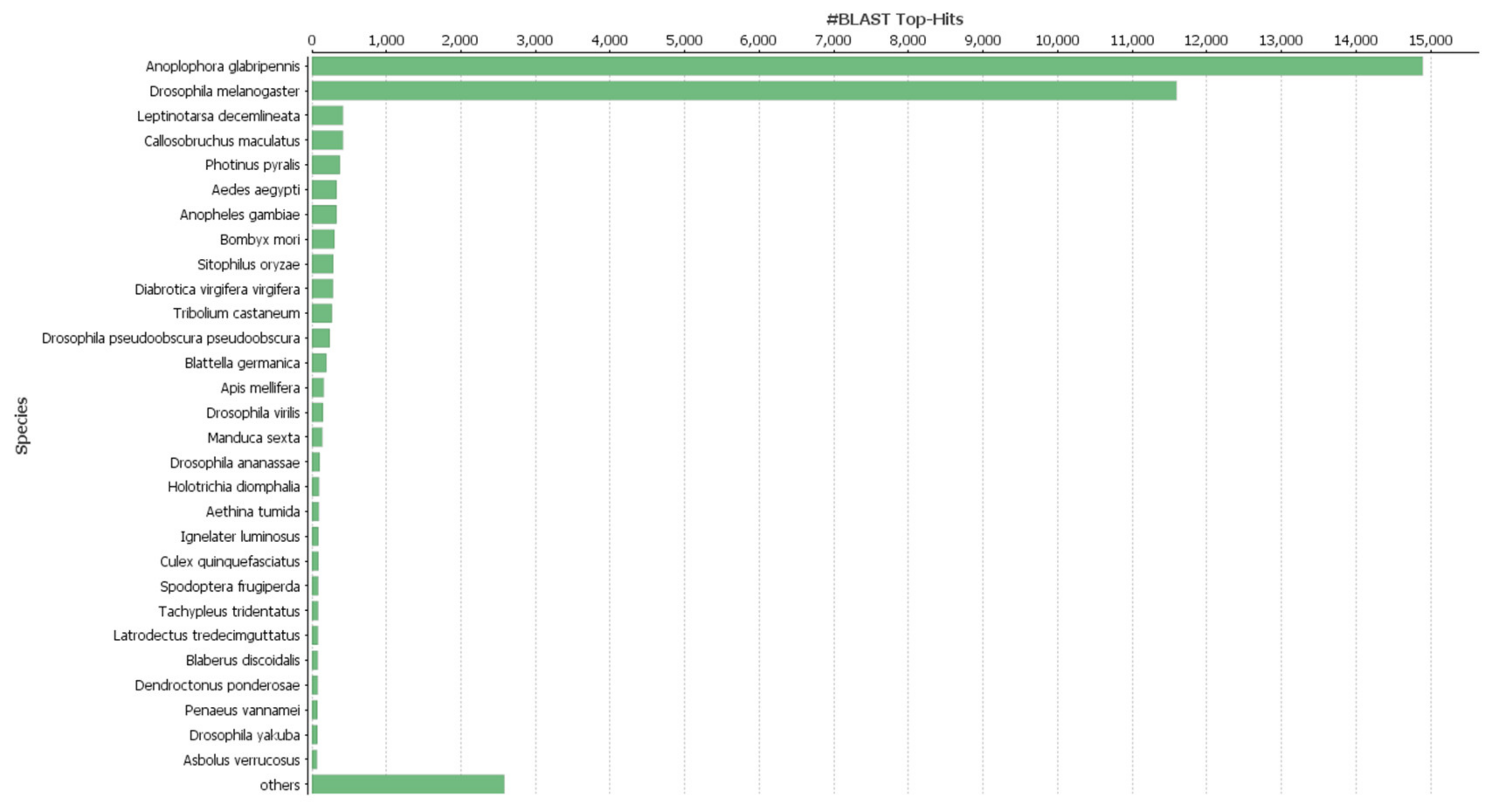
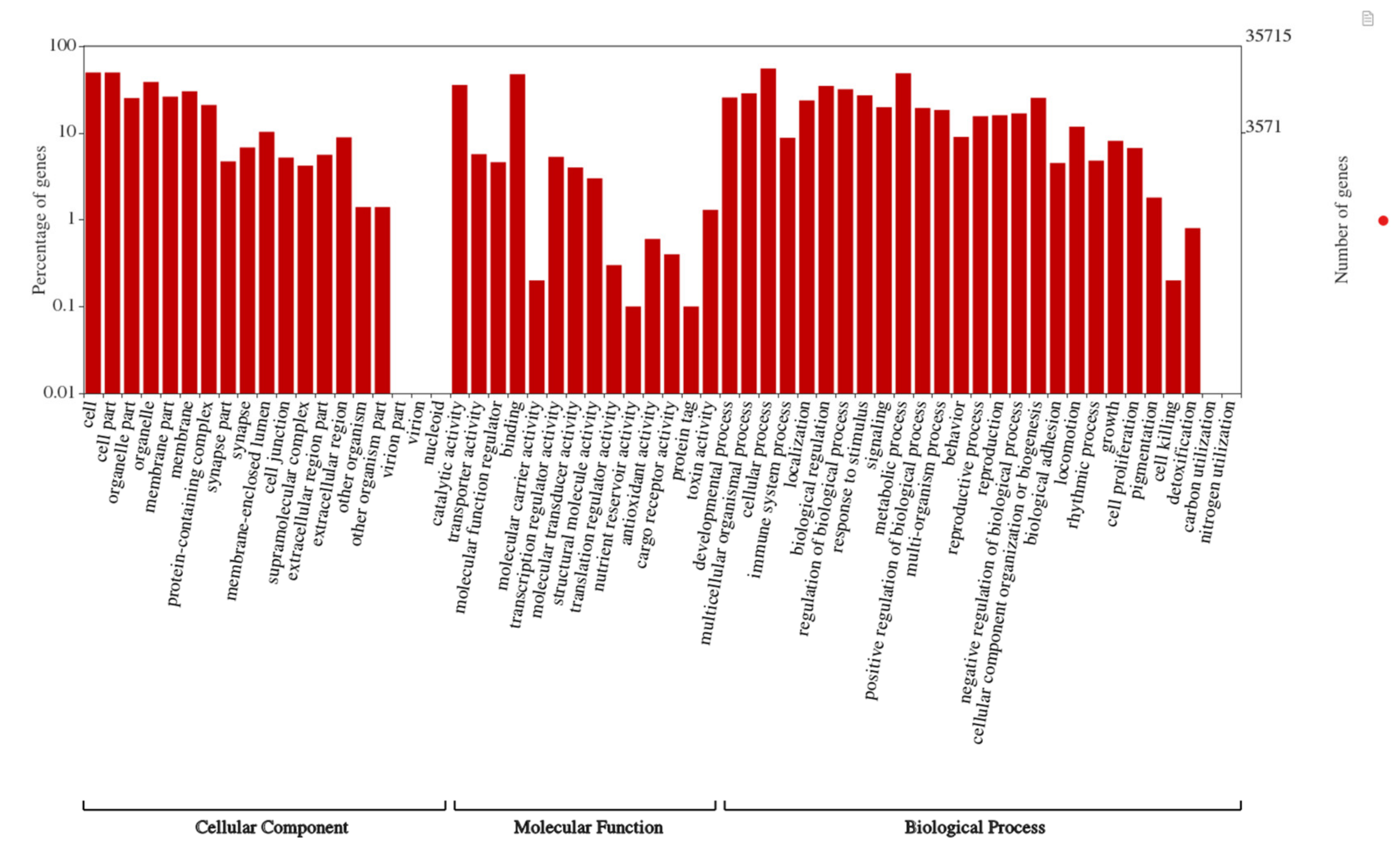
3.2. Functional Annotations, Species Distribution and Differential Gene Expression Analysis (DEG)
3.3. AMP Prediction
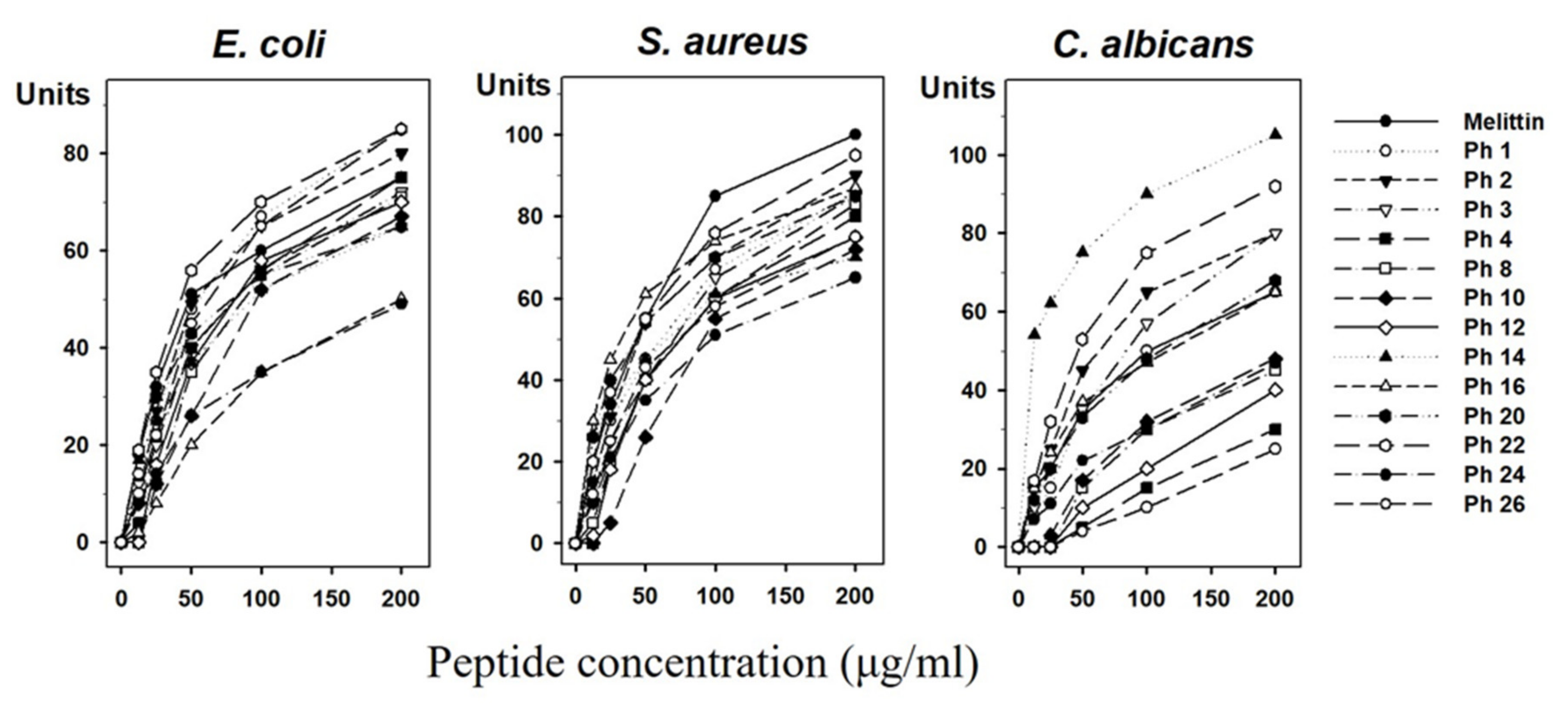
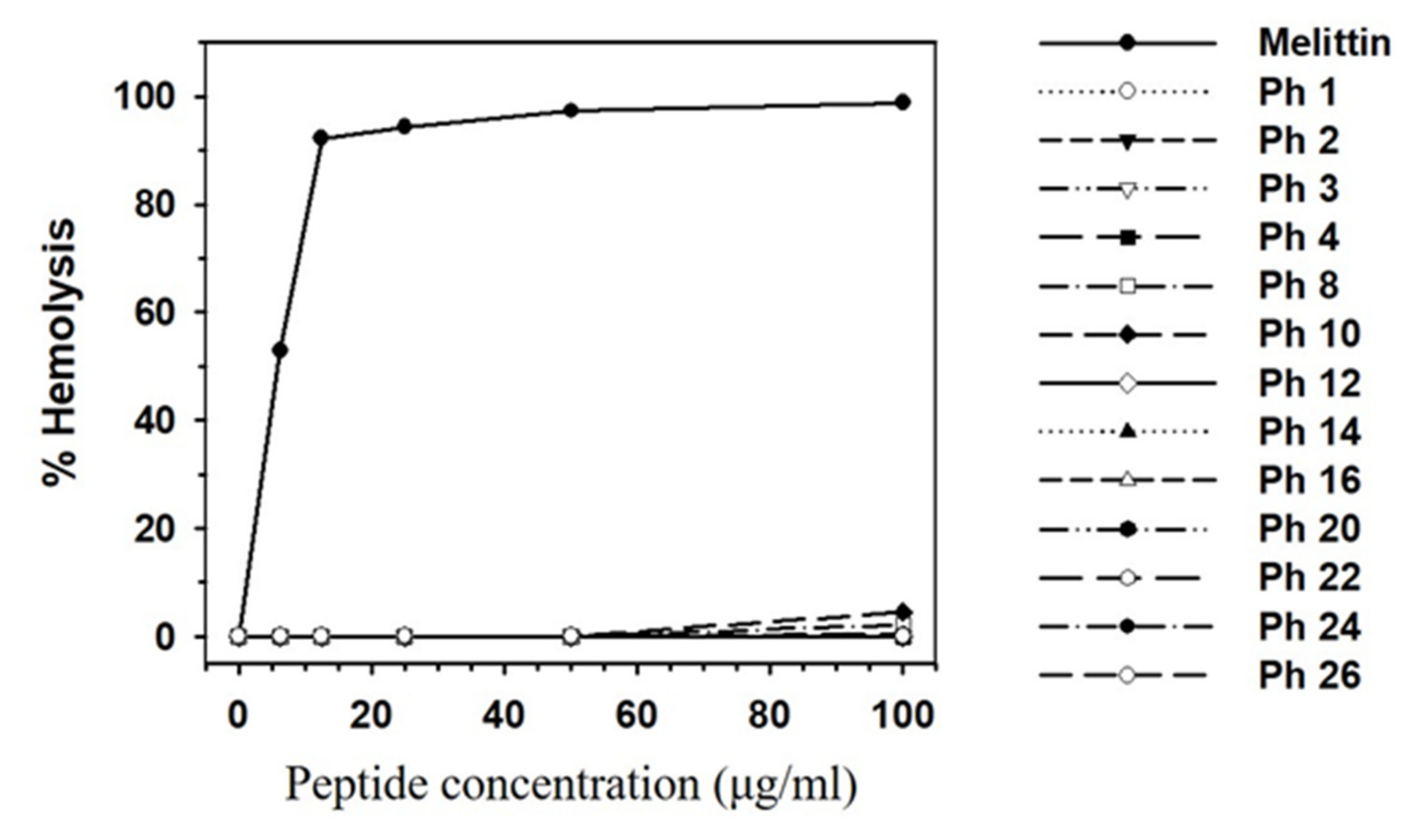
3.4. Experimental Validation of Putative and Novel AMPs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, D.E. Antimicrobial Peptides. Surg. Infect. 2018, 19, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E. Antimicrobial Peptides: An Introduction. Methods Mol. Biol. 2017, 1548, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.; Grapputo, A.; Romoli, O.; Sandrelli, F. Insect Cecropins, Antimicrobial Peptides with Potential Therapeutic Applications. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.Y.; Chowdhury, M.; Huang, Y.D.; Yu, X.Q. Insect antimicrobial peptides and their applications. Appl. Microbiol. Biotechnol. 2014, 98, 5807–5822. [Google Scholar] [CrossRef] [Green Version]
- Tonk, M.; Vilcinskas, A. The Medical Potential of Antimicrobial Peptides from Insects. Curr. Top. Med. Chem. 2017, 17, 554–575. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Kuca, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10. [Google Scholar] [CrossRef]
- Kolmar, H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef]
- Gracy, J.; Chiche, L. Structure and modeling of knottins, a promising molecular scaffold for drug discovery. Curr. Pharm. Des. 2011, 17, 4337–4350. [Google Scholar] [CrossRef]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef]
- Hwang, J.S.; Lee, J.; Hwang, B.; Nam, S.H.; Yun, E.Y.; Kim, S.R.; Lee, D.G. Isolation and characterization of Psacotheasin, a novel Knottin-type antimicrobial peptide, from Psacothea hilaris. J. Microbiol. Biotechnol. 2010, 20, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, B.; Hwang, J.S.; Lee, J.; Lee, D.G. Antifungal properties and mode of action of psacotheasin, a novel knottin-type peptide derived from Psacothea hilaris. Biochem. Biophys. Res. Commun. 2010, 400, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Hwang, J.S.; Lee, J.; Lee, D.G. The antimicrobial peptide, psacotheasin induces reactive oxygen species and triggers apoptosis in Candida albicans. Biochem. Biophys. Res. Commun. 2011, 405, 267–271. [Google Scholar] [CrossRef]
- Yoo, W.G.; Lee, J.H.; Shin, Y.; Shim, J.Y.; Jung, M.; Kang, B.C.; Oh, J.; Seong, J.; Lee, H.K.; Kong, H.S.; et al. Antimicrobial peptides in the centipede Scolopendra subspinipes mutilans. Funct. Integr. Genomics 2014, 14, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.W.; Lee, J.H.; Subramaniyam, S.; Yun, E.Y.; Kim, I.; Park, J.; Hwang, J.S. De Novo Transcriptome Analysis and Detection of Antimicrobial Peptides of the American Cockroach Periplaneta americana (Linnaeus). PLoS ONE 2016, 11, e0155304. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.W.; Markkandan, K.; Lee, J.H.; Subramaniyam, S.; Yoo, S.; Park, J.; Hwang, J.S. Transcriptome Profiling and In Silico Analysis of the Antimicrobial Peptides of the Grasshopper Oxya chinensis sinuosa. J. Microbiol. Biotechnol. 2016, 26, 1863–1870. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.T.; Lee, C.C.; Yang, J.R.; Lai, J.Z.; Chang, K.Y. A large-scale structural classification of antimicrobial peptides. Biomed Res. Int. 2015, 2015, 475062. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic. Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghu, F.H.; Gopi, L.; Barai, R.S.; Ramteke, P.; Nizami, B.; Idicula-Thomas, S. CAMP: Collection of sequences and structures of antimicrobial peptides. Nucleic. Acids Res. 2014, 42, D1154–D1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, D.A.; Lehrer, R.I. Designer assays for antimicrobial peptides. Disputing the “one-size-fits-all” theory. Methods Mol. Biol. 1997, 78, 169–186. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Raw Reads | Size (GB) | Clean Reads | De Novo Alignment (%) |
|---|---|---|---|---|
| PH_N1 1 | 22,668,170 | 6 | 22,247,657 | 81.36 |
| PH_N2 1 | 23,223,996 | 6.9 | 22,706,277 | 81.48 |
| PH_N3 1 | 22,065,414 | 7.3 | 21,617,382 | 81.16 |
| PH_I1 2 | 19,915,844 | 6.8 | 19,547,003 | 86.32 |
| PH_I2 2 | 22,838,132 | 7 | 22,388,341 | 85.36 |
| PH_I3 2 | 24,414,115 | 6.6 | 23,931,670 | 83.60 |
| De novo assembly Unigenes Base pairs Average length of unigenes GC percentage Annotation Descriptions BLAST- Swissprot No blast Gene Ontology | 35,715 73,299,234 2052 37.53 No of hits 33,932 1783 26,492 | Percentage 95.0 5.0 74.0 |
| Propensity | Methods | Cutoff | No. of Peptide Sequences |
|---|---|---|---|
| Raw Sequence | Total peptides (2 to 50 amino acids) | 104,615 | |
| Molecular | Pepstats Pepstats AMPA | Charge > 0 (+) 8 ≥ pI ≤ 12 Stretch no ≥1 | 70,364 51,494 83,783 |
| Aggregation (Invivo) | Tango Tango Tango | AGG (≤500) Helix (≥0 helix ≤25) Beta (≥25 beta ≤100) | 85,941 98,871 48,068 |
| Aggregation (Invitro) | Aggrescan | Na4VSS (≥−40 Na4vSS ≤60) | 80,821 |
| Similarity | BlastP (E value: 1E-05) | Known AMPs (ADAM, >80) Known AMPs (APD, >80) Known AMPs (CAMP, >80) Novel AMPs (Similarity <80) | 0 0 0 14,405 |
| AMP | CAMP CAMP CAMP CAMP ADAM | SVM (>0.5, AMP) RF (>0.5, AMP) ANN (AMP) DA (>0.5, AMP) SVM (>0.5, AMP) | 4653 5019 7363 5655 10,590 |
| Final | 2290 |
| Peptide | Protein Length | CAMP-SVM Score | CAMP-RF Score | CAMP-DA Score | ADAM-SVM Class | Peptide Sequence |
|---|---|---|---|---|---|---|
| Ph 1 | 16 | 0.793 | 0.8945 | 0.732 | 1.63 | RAIKWPGNGLLFLKY * |
| Ph 2 | 15 | 0.701 | 0.976 | 0.959 | 1.84 | KLPIVNVKLVNRIK * |
| Ph 3 | 14 | 0.775 | 0.688 | 0.784 | 1.2 | KRGYQVPRIAFII * |
| Ph 4 | 16 | 0.674 | 0.6385 | 0.965 | 1.2 | KLQVVPAIHLVWLQK * |
| Ph 8 | 14 | 0.832 | 0.7725 | 0.746 | 1.63 | RCLKTCFLSFIRY * |
| Ph 10 | 13 | 0.555 | 0.816 | 0.855 | 0.45 | RISCVAMRLILK * |
| Ph 12 | 13 | 0.551 | 0.6325 | 0.902 | 2.53 | RLLLLCYACGKS * |
| Ph 14 | 10 | 1 | 0.627 | 0.989 | 3.49 | RRRCRCCRY * |
| Ph 16 | 10 | 0.999 | 0.6585 | 0.833 | 2.15 | RKSWRHWKC * |
| Ph 20 | 13 | 0.667 | 0.6365 | 0.751 | 1.2 | KMFTKCIRYRKM * |
| Ph 22 | 12 | 0.93 | 0.621 | 0.554 | 1.06 | KRIFYLYIRGQ * |
| Ph24 | 14 | 0.706 | 0.7095 | 0.595 | 1.56 | KLSNAVFKSCRKI * |
| Ph26 | 14 | 0.898 | 0.534 | 0.788 | 0.72 | KIGTFIKKLSYTS * |
| Peptides (200 μg/mL) | E. coli | S. aureus | C. albicans |
|---|---|---|---|
| Melittin | 75 | 100 | 65 |
| Ph 1 | 85 | 86 | 65 |
| Ph 2 | 80 | 90 | 80 |
| Ph 3 | 72 | 85 | 80 |
| Ph 4 | 75 | 80 | 30 |
| Ph 8 | 71 | 83 | 45 |
| Ph 10 | 67 | 72 | 48 |
| Ph 12 | 70 | 75 | 40 |
| Ph 14 | 65 | 70 | 105 |
| Ph 16 | 50 | 87 | 65 |
| Ph 20 | 65 | 85 | 68 |
| Ph 22 | 85 | 95 | 92 |
| Ph 24 | 49 | 65 | 47 |
| Ph 26 | 85 | 75 | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Chung, H.; Shin, Y.P.; Kim, I.-W.; Natarajan, S.; Veerappan, K.; Seo, M.; Park, J.; Hwang, J.S. Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction. Insects 2020, 11, 676. https://doi.org/10.3390/insects11100676
Lee JH, Chung H, Shin YP, Kim I-W, Natarajan S, Veerappan K, Seo M, Park J, Hwang JS. Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction. Insects. 2020; 11(10):676. https://doi.org/10.3390/insects11100676
Chicago/Turabian StyleLee, Joon Ha, Hoyong Chung, Yong Pyo Shin, In-Woo Kim, Sathishkumar Natarajan, Karpagam Veerappan, Minchul Seo, Junhyung Park, and Jae Sam Hwang. 2020. "Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction" Insects 11, no. 10: 676. https://doi.org/10.3390/insects11100676
APA StyleLee, J. H., Chung, H., Shin, Y. P., Kim, I. -W., Natarajan, S., Veerappan, K., Seo, M., Park, J., & Hwang, J. S. (2020). Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction. Insects, 11(10), 676. https://doi.org/10.3390/insects11100676