Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Isolation
2.2. Sequence Analysis and Gene Annotation
2.3. Phylogenetic Analysis
3. Results and Discussion
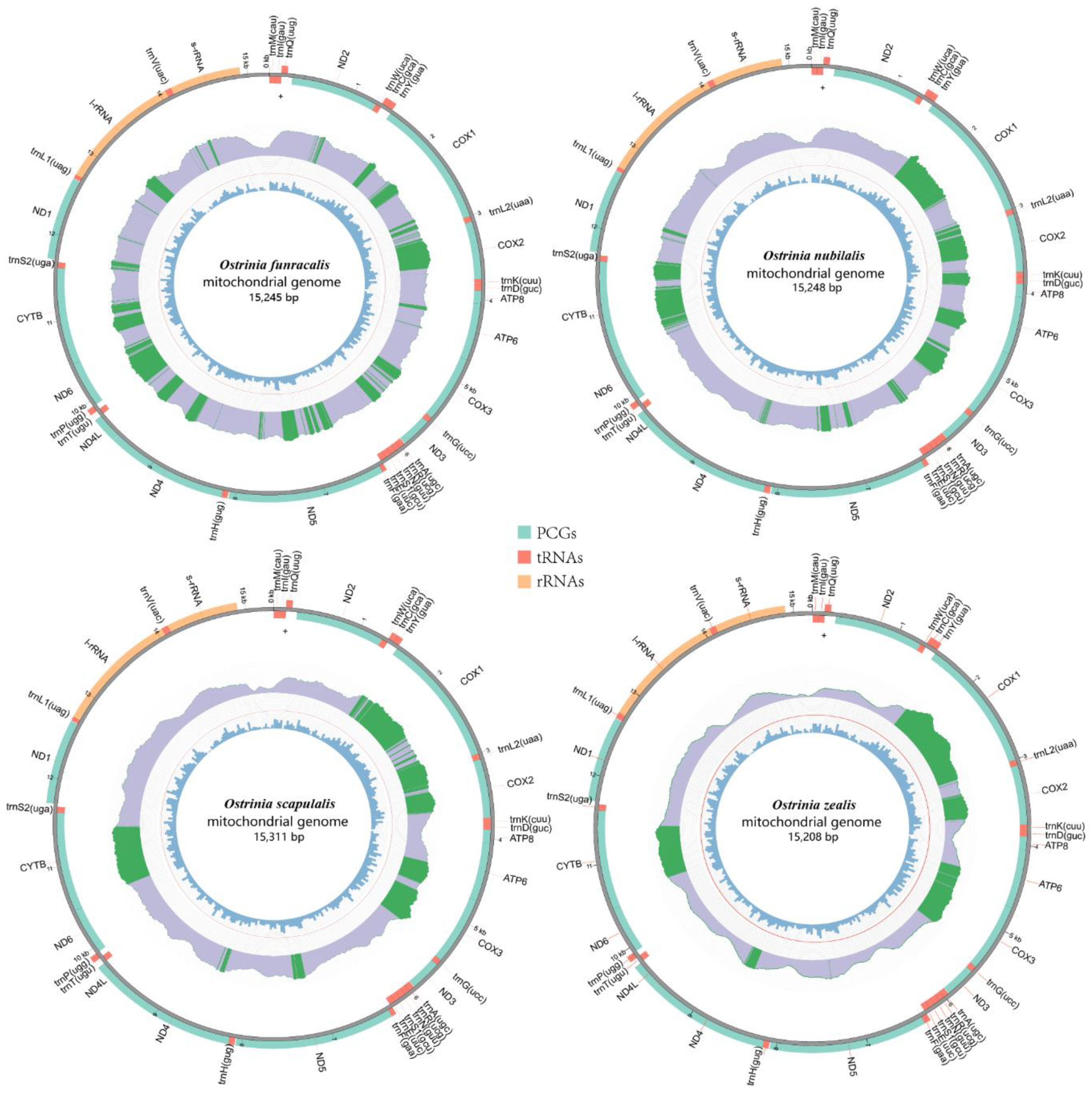
3.1. Mitogenome Structure and Organization
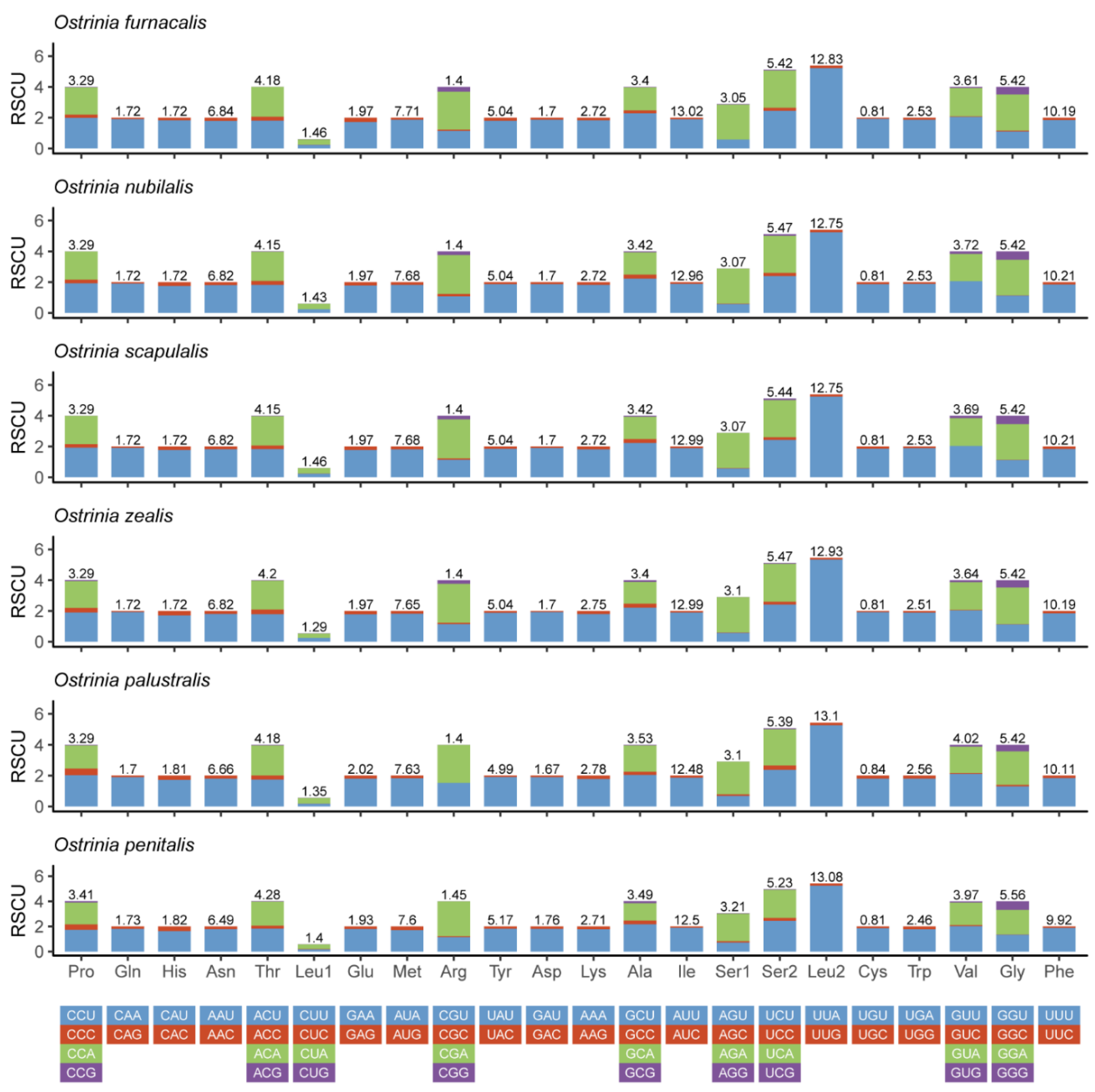
3.2. Protein-coding Genes
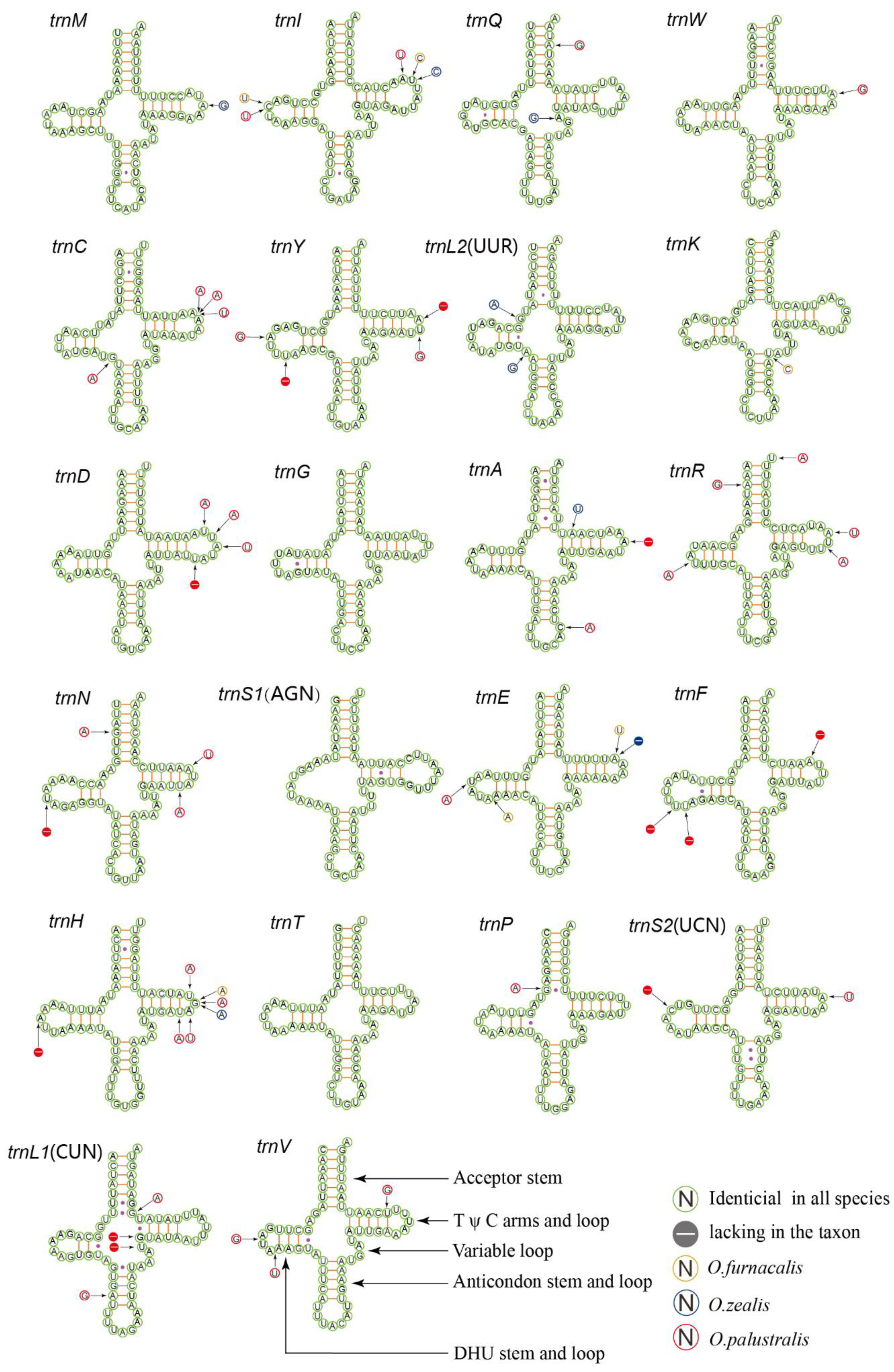
3.3. Ribosomal and Transfer RNA Genes
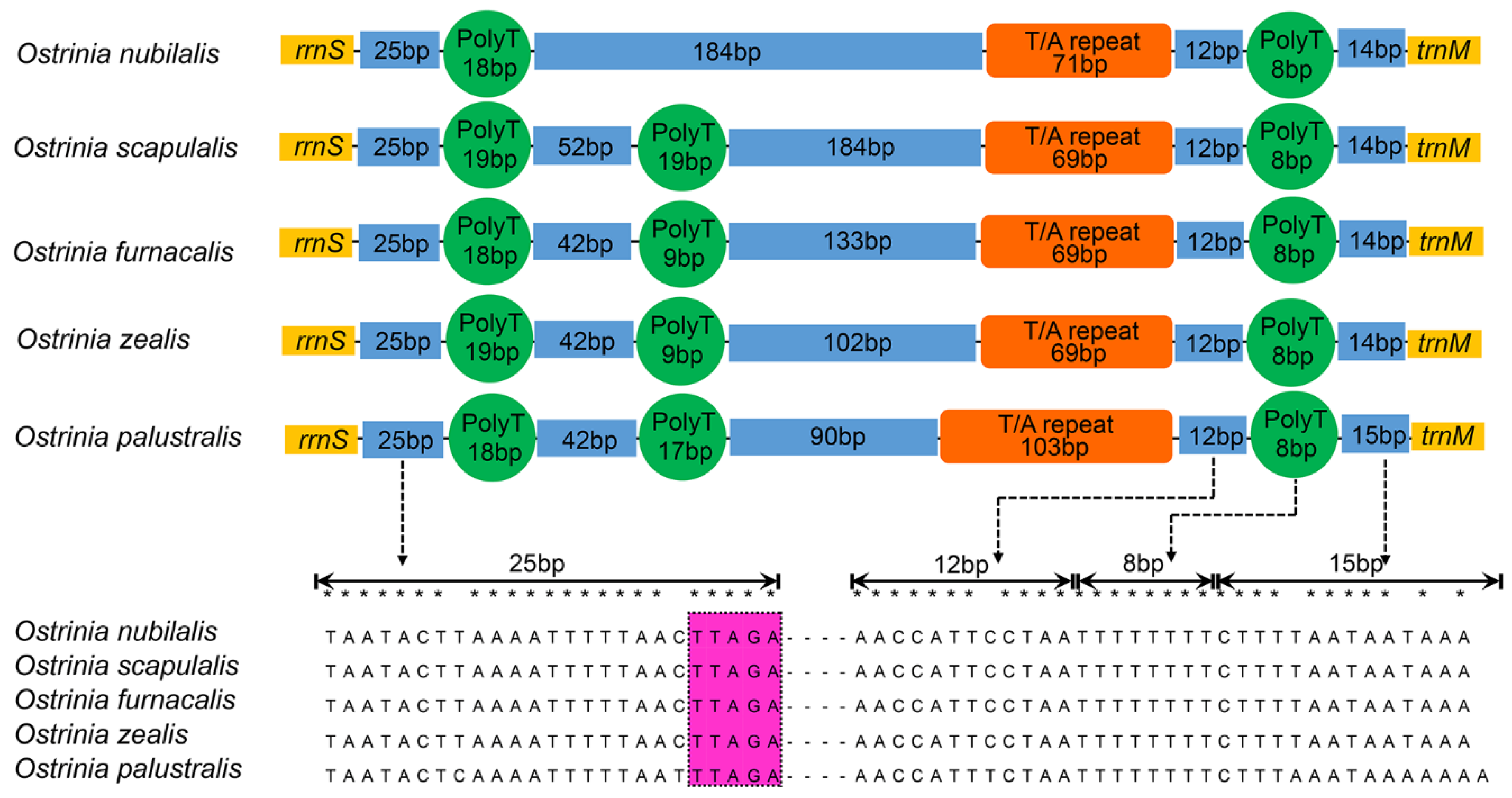
3.4. Control Region
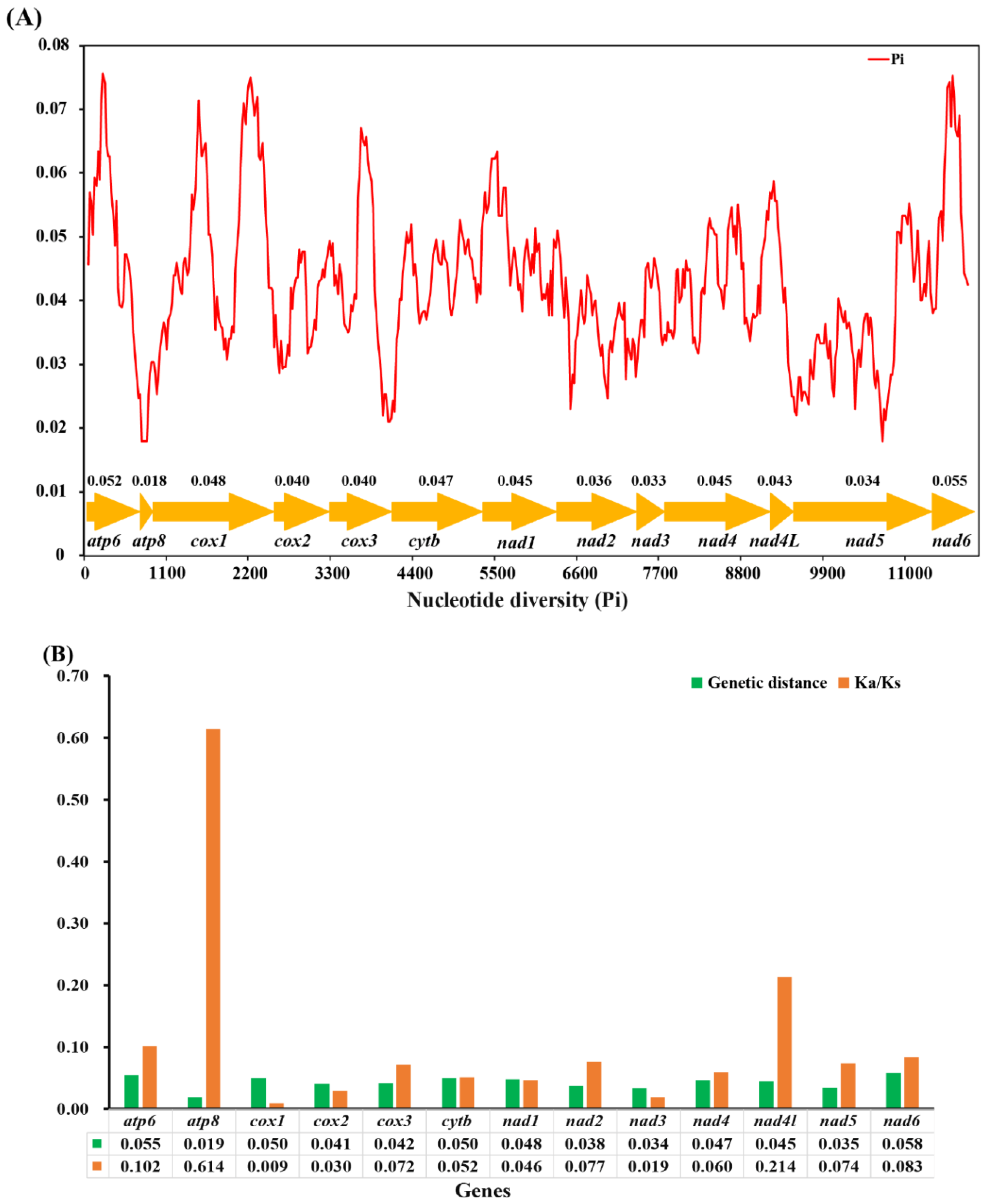
3.5. Nucleotide Diversity Analyses
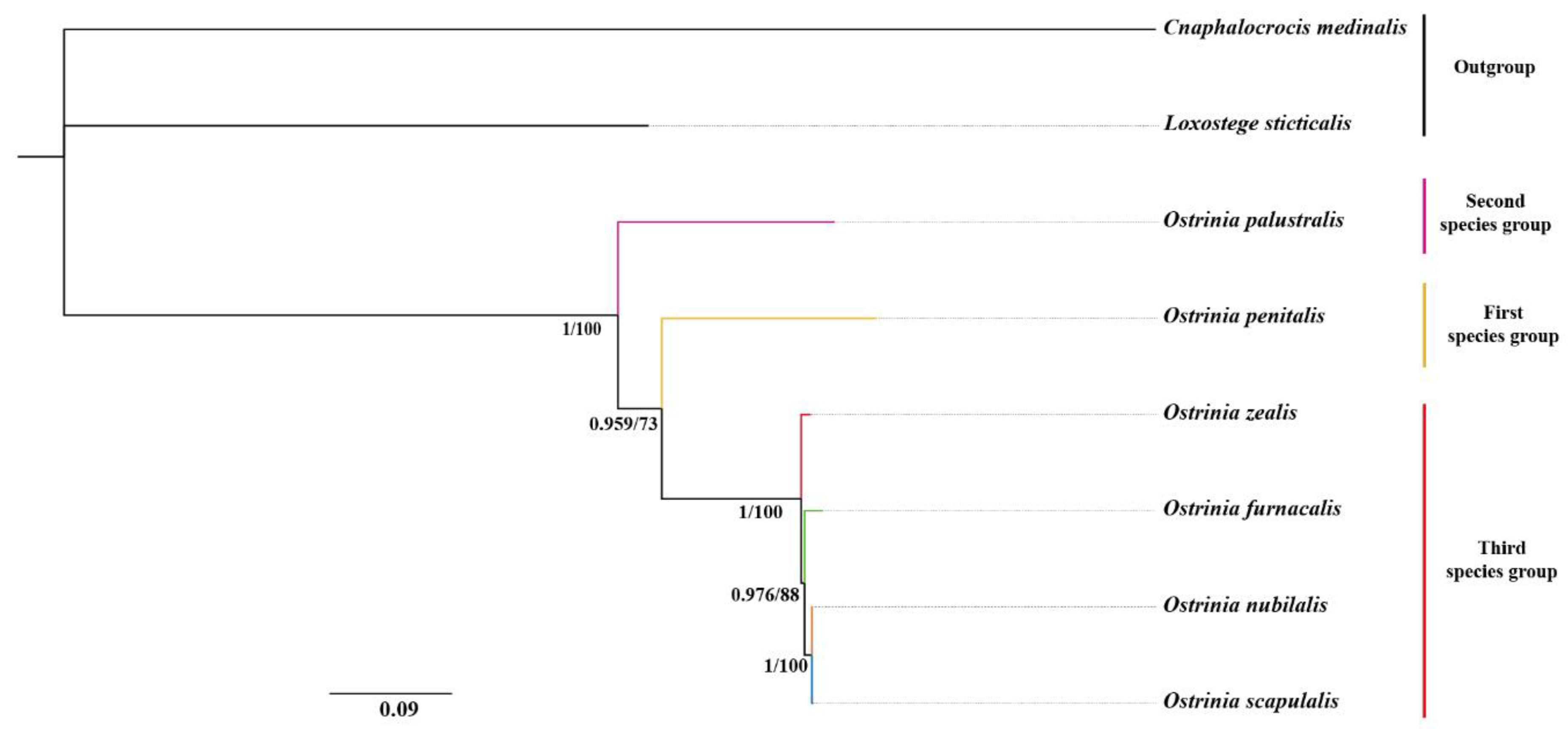
3.6. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, R.S.; Wang, Z.Y.; He, K.L. Advances in phylogenetic and taxonomic studies on genus Ostrinia. Plant Prot. Beijing 2007, 33, 20–25. [Google Scholar] [CrossRef]
- Mutuura, A.; Munroe, E. Taxonomy and distribution of the European corn borer and allied species: Genus Ostrinia (Lepidoptera: Pyralidae). Mem. Entomol. Soc. Can. 1970, 102, 1–112. [Google Scholar] [CrossRef]
- Park, K.T. Taxonomic study of the corn stem borer in Korea with allied species of the genus Ostrinia (Lep.; Pyralidae). Korean. J. Appl. Entomol. 1975, 14, 221–225. [Google Scholar]
- Jiang, F.; Zhang, T.; Bai, S.; Wang, Z.; He, K. Evaluation of Bt corn with pyramided genes on efficacy and insect resistance management for the Asian corn borer in China. PLoS ONE 2016, 11, e0168442. [Google Scholar] [CrossRef] [Green Version]
- Afidchao, M.M.; Musters, C.J.; de Snoo, G.R. Asian corn borer (ACB) and non-ACB pests in GM corn (Zea mays L.) in the Philippines. Pest Manag. Sci. 2013, 69, 792–801. [Google Scholar] [CrossRef]
- Zhang, F.; Babendreier, D.; Wang, Z.Y.; Il, K.S.; Zheng, L.; Pyon, Y.C.; Bai, S.X.; Song, K.; Ri, J.O.; Grossrieder, M.; et al. Mass releases of Trichogramma ostriniae increase maize production in DPR Korea. J. Appl. Entomol. 2010, 134, 481–490. [Google Scholar] [CrossRef]
- Wu, L.H.; Hill, M.P.; Thomson, L.J.; Hoffmann, A.A. Assessing the current and future biological control potential of Trichogramma ostriniae on its hosts Ostrinia furnacalis and Ostrinia nubilalis. Pest Manag. Sci. 2018, 74, 1513–1523. [Google Scholar] [CrossRef]
- Bourguet, D.; Chaufaux, J.; Seguin, M.; Buisson, C.; Hinton, J.L.; Stodola, T.J.; Porter, P.; Cronholm, G.; Buschman, L.L.; Andow, D.A. Frequency of alleles conferring resistance to Bt maize in French and US corn belt populations of the European corn borer, Ostrinia nubilalis. Theor. Appl. Genet. 2003, 106, 1225–1233. [Google Scholar] [CrossRef]
- Gemeno, C.; Sans, A.; Lopez, C.; Albajes, R.; Eizaguirre, M. Pheromone antagonism in the European corn borer moth Ostrinia nubilalis. J. Chem. Ecol. 2006, 32, 1071–1084. [Google Scholar] [CrossRef]
- Frolov, A.N. Genetic analysis of ‘large’ tibia—The taxonomic character of brush-leg borer Ostrinia scapulalis Wlk. (Lepidoptera, Pyraustidae). Genetika 1981, 17, 2160–2166. (In Russian) [Google Scholar]
- Frolov, A.N. Biotaxonomic analysis of harmful species of the genus Ostrinia Hbn. Ethologiya Nasek. Tr. Vsesoyuznogo Entomol. Obs. 1984, 6, 4–100. (In Russian) [Google Scholar]
- Frolov, A.N.; Audiot, P.; Bourguet, D.; Kononchuk, A.G.; Malysh, J.M.; Ponsard, S.; Streiff, R.; Tokarev, Y.S. From Russia with lobe: Genetic differentiation in trilobed uncus Ostrinia spp. Follows food plant, not hairy legs. Heredity 2012, 108, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolov, A.N.; Bourguet, D.; Ponsard, S. Reconsidering the taxomony of several Ostrinia species in the light of reproductive isolation: A tale for Ernst Mayr. Biol. J. Linn. Soc. 2007, 91, 49–72. [Google Scholar] [CrossRef] [Green Version]
- Frolov, A.N. Variation in the European corn borer, Ostrinia nubilalis, and allies (Lepidoptera: Pyralidae). Mem. Soc. R. Belg. Entomol. 1998, 38, 1–21. [Google Scholar]
- Kim, C.G.; Hoshizaki, S.; Huang, Y.P.; Tatsuki, S.; Ishikawa, Y. Usefulness of mitochondrial COII gene sequences in examining phylogenetic relationships in the Asian corn borer, Ostrinia furnacalis, and allied species (Lepidoptera: Pyralidae). Appl. Entomol. Zool. 1999, 34, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.S.; Wang, Z.Y.; He, K.L. Genetic diversity and phylogeny of the genus Ostrinia (Lepidoptera: Crambidae) inhabiting China inferred from mitochondrial COI gene. J. Nanjing Agric. Univ. 2011, 34, 73–80. [Google Scholar]
- Lassance, J.M.; Lienard, M.A.; Antony, B.; Qian, S.; Fujii, T.; Tabata, J.; Ishikawa, Y.; Lofstedt, C. Functional consequences of sequence variation in the pheromone biosynthetic gene pgFAR for Ostrinia moths. Proc. Natl. Acad. Sci. India Sect. A 2013, 110, 3967–3972. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef] [Green Version]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Simon, C. Molecular systematics at the species boundary: Exploiting conserved and variable regions of the mitochondrial genome of animals via direct sequencing from amplified DNA. In Molecular Techniques in Taxonomy; Springer: Berlin/Heidelberg, Germany, 1991; pp. 33–71. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Chen, M.Y.; Wang, J.F.; Liang, A.P.; Lin, C.P. Some mitochondrial genes perform better for damselfly phylogenetics: Species-and population-level analyses of four complete mitogenomes of Euphaea sibling species. Syst. Entomol. 2018, 43, 702–715. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, Y.; Zhou, X.; Kong, X.; Wei, S.; Ward, R.D.; Zhang, A.-B. Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes. BMC Genom. 2015, 16, 428. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol. Evol. 2006, 21, 439–446. [Google Scholar] [CrossRef]
- Hwang, E.J.; Kim, M.J.; Kim, S.S.; Kim, I. Complete mitochondrial genome of Ostrinia palustralis memnialis Walker, 1859 (Lepidoptera: Crambidae). Mitochondrial DNA Part B 2019, 4, 1364–1366. [Google Scholar] [CrossRef]
- Coates, B.S.; Sumerford, D.V.; Hellmich, R.L.; Lewis, L.C. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int. J. Biol. Sci. 2005, 1, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Coates, B.S.; Abel, C.A. The mitochondrial genome of the American lotus borer, Ostrinia penitalis (Lepidoptera: Crambidae). Mitochondrial DNA Part A 2016, 27, 1938–1939. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. India Sect. A 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [Green Version]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.L.; Kim, M.J.; Kim, I. Description of new mitochondrial genomes (Spodoptera litura, Noctuoidea and Cnaphalocrocis medinalis, Pyraloidea) and phylogenetic reconstruction of Lepidoptera with the comment on optimization schemes. Mol. Biol. Rep. 2013, 40, 6333–6349. [Google Scholar] [CrossRef]
- Ma, H.F.; Zheng, X.X.; Peng, M.H.; Bian, H.X.; Chen, M.M.; Liu, Y.Q.; Jiang, X.; Qin, L. Complete mitochondrial genome of the meadow moth, Loxostege sticticalis (Lepidoptera: Pyraloidea: Crambidae), compared to other Pyraloidea moths. J. Asia Pac. Entomol. 2016, 19, 697–706. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Kang, A.R.; Jeong, H.C.; Kim, K.G.; Kim, I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol. Phylogenet. Evol. 2011, 61, 436–445. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Jiang, X.H.; Hou, X.H.; Yang, H.; Chen, W.L. The mitochondrial genome of Ephestia elutella (Insecta: Lepidoptera: Pyralidae). Mitochondrial DNA Part B 2018, 3, 189–190. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, M.J.; Ahn, S.J.; Kim, I. Complete mitochondrial genome of the grass moth Glyphodes quadrimaculalis (Lepidoptera: Crambidae). Mitochondrial DNA Part A 2015, 26, 247–249. [Google Scholar] [CrossRef]
- Yang, M.S.; Shi, S.S.; Dai, P.; Song, L.; Liu, X.M. Complete mitochondrial genome of Palpita hypohomalia (Lepidoptera: Pyraloidea: Crambidae) and its phylogenetic implications. Eur. J. Entomol. 2018, 115, 708–717. [Google Scholar] [CrossRef]
- Yang, M.S.; Song, L.; Mao, J.H.; Shi, Y.X.; Wu, C.J.; Zhang, Y.X.; Huang, L.; Peng, W.F.; Liu, X.M. Complete mitochondrial genome of the soybean leaffolder, Omiodes indicata (Lepidoptera: Pyraloidea: Crambidae), and phylogenetic analysis for Pyraloidea. Int. J. Biol. Macromol. 2018, 115, 53–60. [Google Scholar] [CrossRef]
- Dai, L.S.; Zhou, X.D.; Kausar, S.; Abbas, M.N.; Wu, L.; Zhou, H.L. Mitochondrial genome of Diaphania indica(saunders) (Lepidoptera: Pyraloidea) and implications for its phylogeny. Int. J. Biol. Macromol. 2018, 108, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.N.; Chai, X.Y.; Bian, D.D.; Zhou, C.L.; Tang, B.P. The complete mitochondrial genome of Plodia interpunctella (Lepidoptera: Pyralidae) and comparison with other Pyraloidea insects. Genome 2016, 59, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yan, J.; Song, J.; You, P. The first mitochondrial genomes for Pyralinae (Pyralidae) and Glaphyriinae (Crambidae), with phylogenetic implications of Pyraloidea. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.F.; McKechnie, S.W.; Pierce, N.; Kreitman, M. The lepidopteran mitochondrial control region: Structure and evolution. Mol. Biol. Evol. 1993, 10, 1259–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Chen, S.; Li, F.H.; Lan, X.E.; You, P. The complete mitochondrial genome of Pycnarmon lactiferalis (Lepidoptera: Crambidae). Mitochondrial DNA Part B 2016, 1, 638–639. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.N.; Du, Y.Z.; Zhai, B.P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci. 2012, 8, 561–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.S.; Du, Y.Z. Characterization of the complete mitochondrial genome of Chilo auricilius and comparison with three other rice stem borers. Gene 2014, 548, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, F.; Chiba, H.; Yuan, X. The mitochondrial genomes of three skippers: Insights into the evolution of the family Hesperiidae (Lepidoptera). Genomics 2019, 112, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Lunt, D.H.; Zhang, D.X.; Szymura, J.M.; Hewltt, O.M. The insect cytochrome oxidase I gene: Evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol. Biol. 1996, 5, 153–165. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Whole Length | GenBank Accession No. | Reference |
|---|---|---|---|---|
| Pyraustinae | Loxostege sticticalis | 15,218 | KR080490 | [41] |
| Ostrinia palustralis | 15,246 | MH574940 | [28] | |
| Ostrinia penitalis | 12,612 | KM395814.1 | [30] | |
| Ostrinia furnacalis | 15,245 | MN793323 | This study | |
| Ostrinia nubilalis | 15,248 | MN793322 | This study | |
| Ostrinia scapulalis | 15,311 | MN793324 | This study | |
| Ostrinia zealis | 15,208 | MN793325 | This study | |
| Spilomelinae | Cnaphalocrocis medinalis | 15,368 | NC_022669 | [40] |
| Regions | Species | Size (bp) | T% | C% | A% | G% | AT(%) | GC(%) | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|---|---|---|
| Full genome | O. furnacalis | 15,245 | 39.2 | 11.4 | 41.8 | 7.6 | 81.0 | 19.0 | 0.032 | −0.199 |
| O. nubilalis | 15,248 | 39.2 | 11.5 | 41.7 | 7.7 | 80.9 | 19.2 | 0.031 | −0.196 | |
| O. scapulalis | 15,311 | 39.3 | 11.4 | 41.7 | 7.7 | 81.0 | 19.1 | 0.030 | −0.196 | |
| O. zealis | 15,208 | 39.2 | 11.4 | 41.7 | 7.7 | 80.9 | 19.1 | 0.031 | −0.193 | |
| O. palustralis | 15,246 | 38.8 | 11.6 | 41.8 | 7.8 | 80.6 | 19.4 | 0.036 | −0.198 | |
| PCGs | O. furnacalis | 11,163 | 45.0 | 9.9 | 34.5 | 10.6 | 79.5 | 20.5 | −0.132 | 0.036 |
| O. nubilalis | 11,163 | 45.0 | 9.9 | 34.4 | 10.7 | 79.4 | 20.6 | −0.133 | 0.042 | |
| O. scapulalis | 11,163 | 45.0 | 9.8 | 34.4 | 10.7 | 79.4 | 20.5 | −0.133 | 0.043 | |
| O. zealis | 11,163 | 45.0 | 9.8 | 34.5 | 10.7 | 79.5 | 20.5 | −0.132 | 0.040 | |
| O. palustralis | 11,160 | 44.9 | 10.0 | 34.2 | 10.8 | 79.1 | 20.8 | −0.135 | 0.039 | |
| 1st codon position | O. furnacalis | 3721 | 37.0 | 9.6 | 37.4 | 16.0 | 74.4 | 25.6 | 0.006 | 0.253 |
| O. nubilalis | 3721 | 37.0 | 9.5 | 37.3 | 16.2 | 74.3 | 25.7 | 0.004 | 0.258 | |
| O. scapulalis | 3721 | 37.0 | 9.6 | 37.3 | 16.2 | 74.3 | 25.8 | 0.005 | 0.256 | |
| O. zealis | 3721 | 37.1 | 9.4 | 37.4 | 16.1 | 74.5 | 25.5 | 0.004 | 0.262 | |
| O. palustralis | 3720 | 37.2 | 9.5 | 36.7 | 16.6 | 73.9 | 26.1 | −0.006 | 0.272 | |
| 2nd codon position | O. furnacalis | 3721 | 48.7 | 16.2 | 21.9 | 13.2 | 70.6 | 29.4 | −0.379 | −0.104 |
| O. nubilalis | 3721 | 48.6 | 16.3 | 21.9 | 13.2 | 70.5 | 29.5 | −0.379 | −0.105 | |
| O. scapulalis | 3721 | 48.6 | 16.3 | 21.9 | 13.2 | 70.5 | 29.5 | −0.379 | −0.104 | |
| O. zealis | 3721 | 48.6 | 16.3 | 21.9 | 13.2 | 70.5 | 29.5 | −0.378 | −0.106 | |
| O. palustralis | 3720 | 48.5 | 16.3 | 21.8 | 13.3 | 70.3 | 29.6 | −0.380 | −0.103 | |
| 3rd codon position | O. furnacalis | 3721 | 49.3 | 3.8 | 44.2 | 2.6 | 93.5 | 6.4 | −0.055 | −0.188 |
| O. nubilalis | 3721 | 49.3 | 3.8 | 44.0 | 2.9 | 93.3 | 6.7 | −0.056 | −0.141 | |
| O. scapulalis | 3721 | 49.4 | 3.7 | 44.1 | 2.8 | 93.5 | 6.5 | −0.057 | −0.132 | |
| O. zealis | 3721 | 49.4 | 3.8 | 44.2 | 2.7 | 93.6 | 6.5 | −0.055 | −0.167 | |
| O. palustralis | 3720 | 49.1 | 4.2 | 44.1 | 2.6 | 93.2 | 6.8 | −0.054 | −0.234 | |
| tRNAs | O. furnacalis | 1480 | 39.8 | 7.8 | 41.7 | 10.7 | 81.5 | 18.5 | 0.023 | 0.161 |
| O. nubilalis | 1480 | 39.8 | 7.7 | 41.7 | 10.8 | 81.5 | 18.5 | 0.023 | 0.168 | |
| O. scapulalis | 1479 | 39.8 | 7.7 | 41.6 | 10.8 | 81.4 | 18.5 | 0.022 | 0.168 | |
| O. zealis | 1477 | 39.9 | 7.8 | 41.4 | 10.9 | 81.3 | 18.7 | 0.019 | 0.167 | |
| O. palustralis | 1481 | 39.7 | 7.3 | 42.1 | 10.9 | 81.8 | 18.2 | 0.030 | 0.197 | |
| rRNAs | O. furnacalis | 2120 | 43.3 | 4.8 | 42.0 | 9.9 | 85.3 | 14.7 | −0.016 | 0.350 |
| O. nubilalis | 2118 | 43.3 | 4.8 | 42.0 | 9.9 | 85.3 | 14.7 | −0.015 | 0.350 | |
| O. scapulalis | 2117 | 43.3 | 4.8 | 42.0 | 9.9 | 85.3 | 14.7 | −0.015 | 0.348 | |
| O. zealis | 2117 | 43.3 | 4.8 | 42.0 | 9.9 | 85.3 | 14.7 | −0.014 | 0.344 | |
| O. palustralis | 2110 | 43.5 | 4.9 | 41.2 | 10.4 | 84.7 | 15.3 | −0.027 | 0.362 | |
| Control region | O. furnacalis | 330 | 51.2 | 4.5 | 42.7 | 1.5 | 93.9 | 6.0 | −0.090 | −0.500 |
| O. nubilalis | 332 | 51.2 | 4.5 | 42.5 | 1.8 | 93.7 | 6.3 | −0.093 | −0.429 | |
| O. scapulalis | 402 | 52.5 | 4.5 | 41.3 | 1.7 | 93.8 | 6.2 | −0.119 | −0.440 | |
| O. zealis | 300 | 51.7 | 5.0 | 42.3 | 1.0 | 94.0 | 6.0 | −0.099 | −0.667 | |
| O. palustralis | 330 | 50.3 | 3.3 | 45.5 | 0.9 | 95.8 | 4.2 | −0.051 | −0.571 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Dong, Y.; Qiao, P.; Yang, Z. Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae). Insects 2020, 11, 232. https://doi.org/10.3390/insects11040232
Zhou N, Dong Y, Qiao P, Yang Z. Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae). Insects. 2020; 11(4):232. https://doi.org/10.3390/insects11040232
Chicago/Turabian StyleZhou, Nan, Yanling Dong, Pingping Qiao, and Zhaofu Yang. 2020. "Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae)" Insects 11, no. 4: 232. https://doi.org/10.3390/insects11040232
APA StyleZhou, N., Dong, Y., Qiao, P., & Yang, Z. (2020). Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae). Insects, 11(4), 232. https://doi.org/10.3390/insects11040232