
Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Transcriptome Sample Preparation
2.2. RNA Extraction and SMRT Sequencing
2.3. Functional Annotation and Structure Analysis
2.4. EST-SSR Detection and Primer Design
2.5. Amplification and Validation of EST-SSRs
3. Results
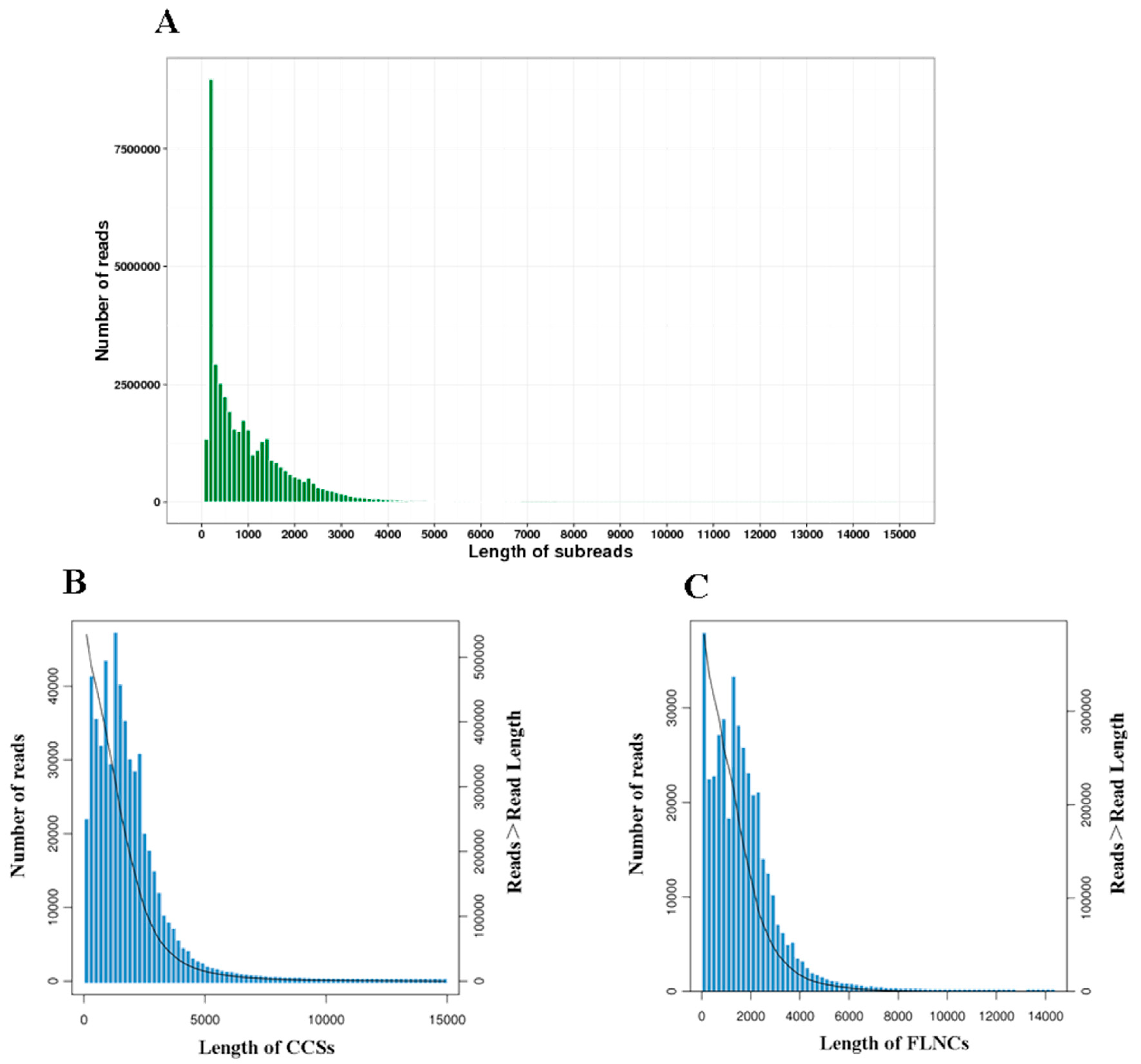
3.1. SMRT Sequencing
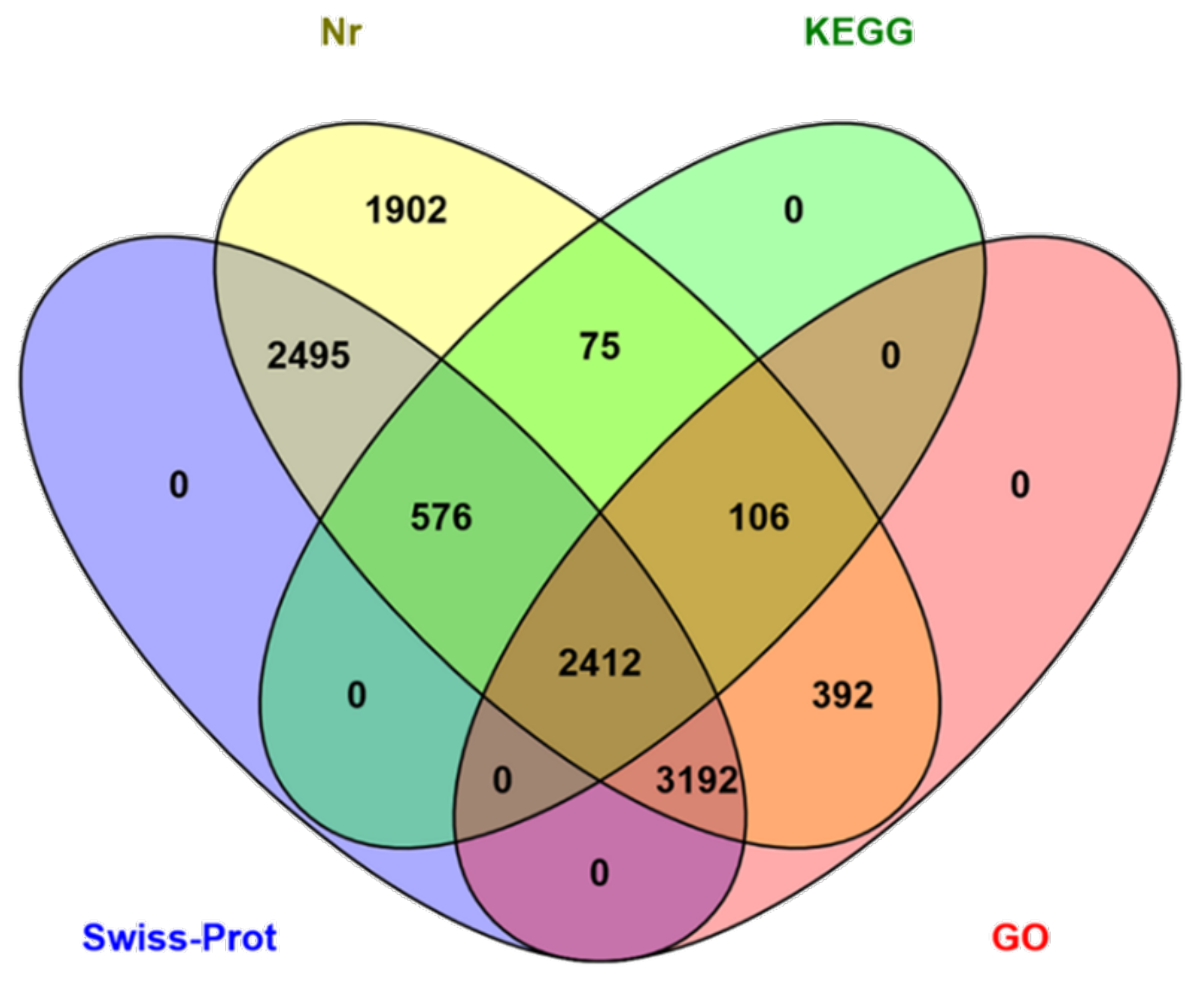
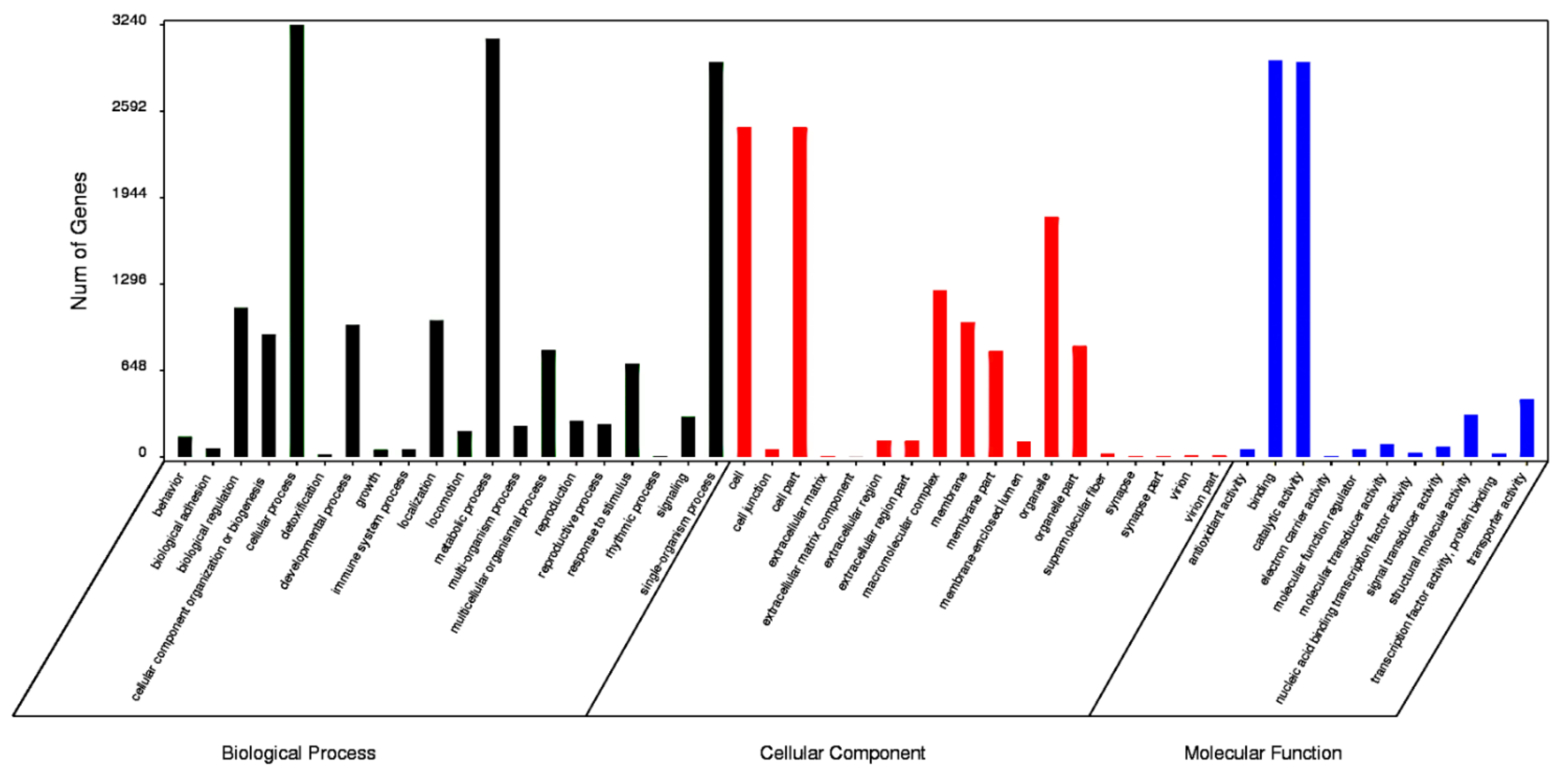
3.2. Functional Annotation
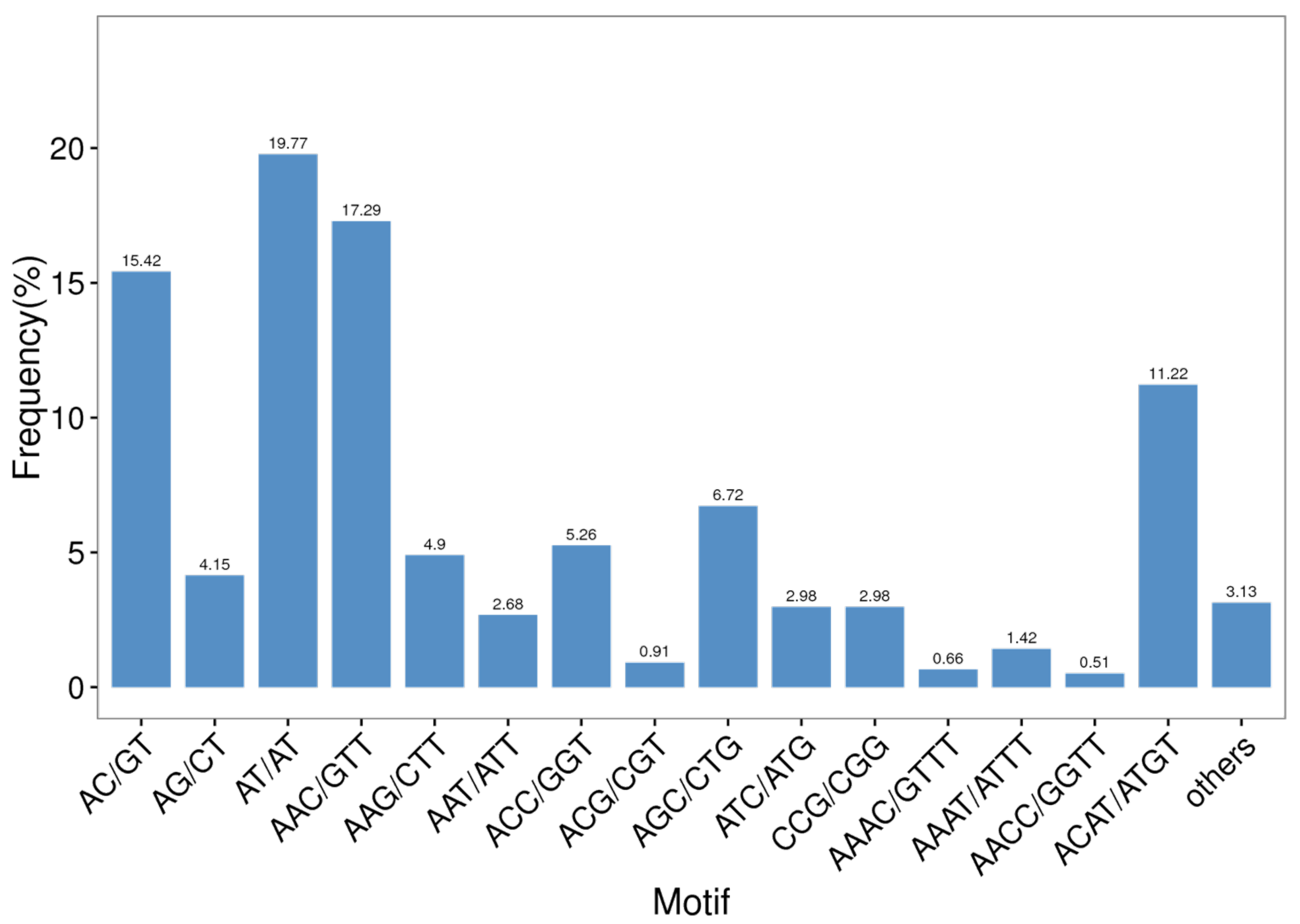
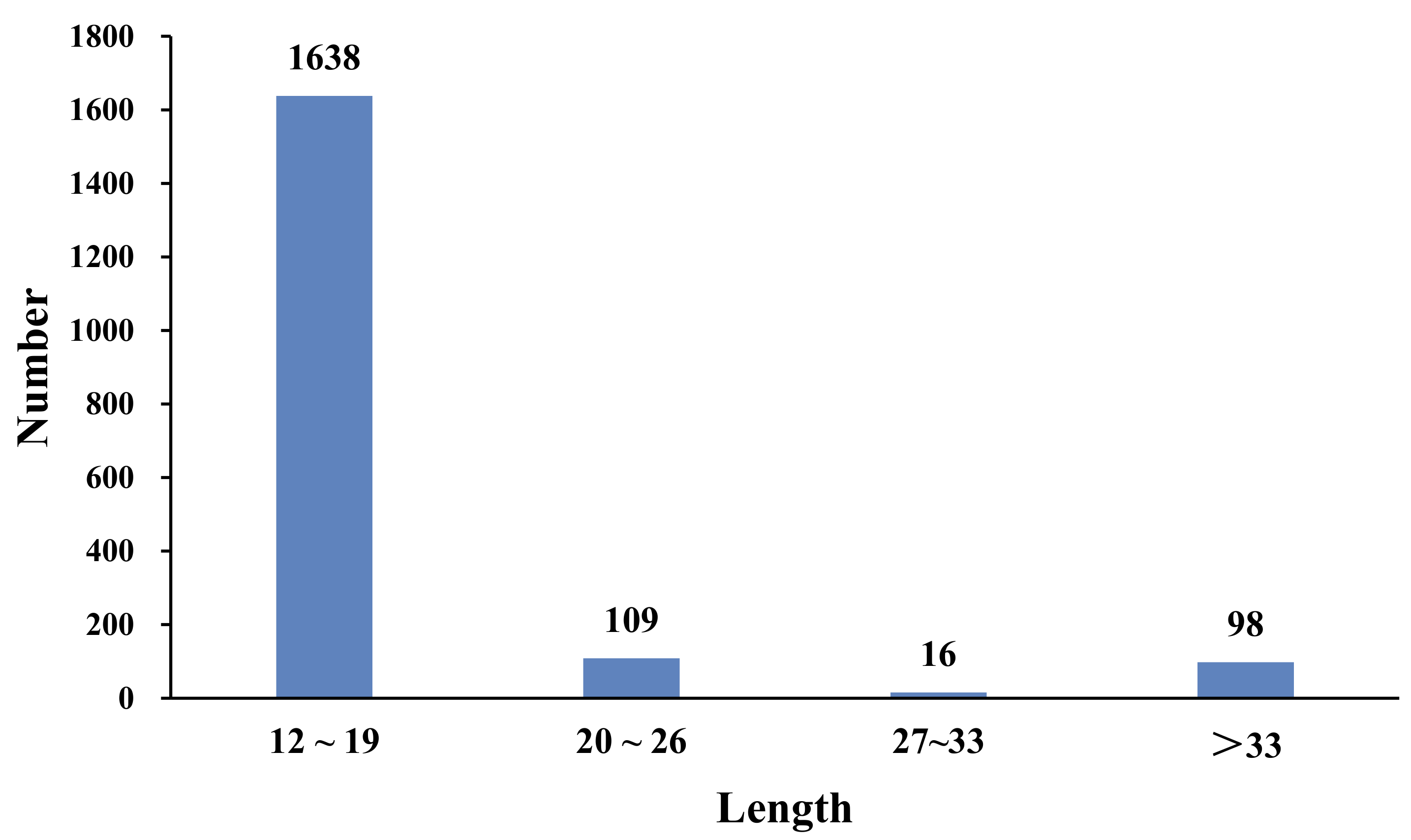
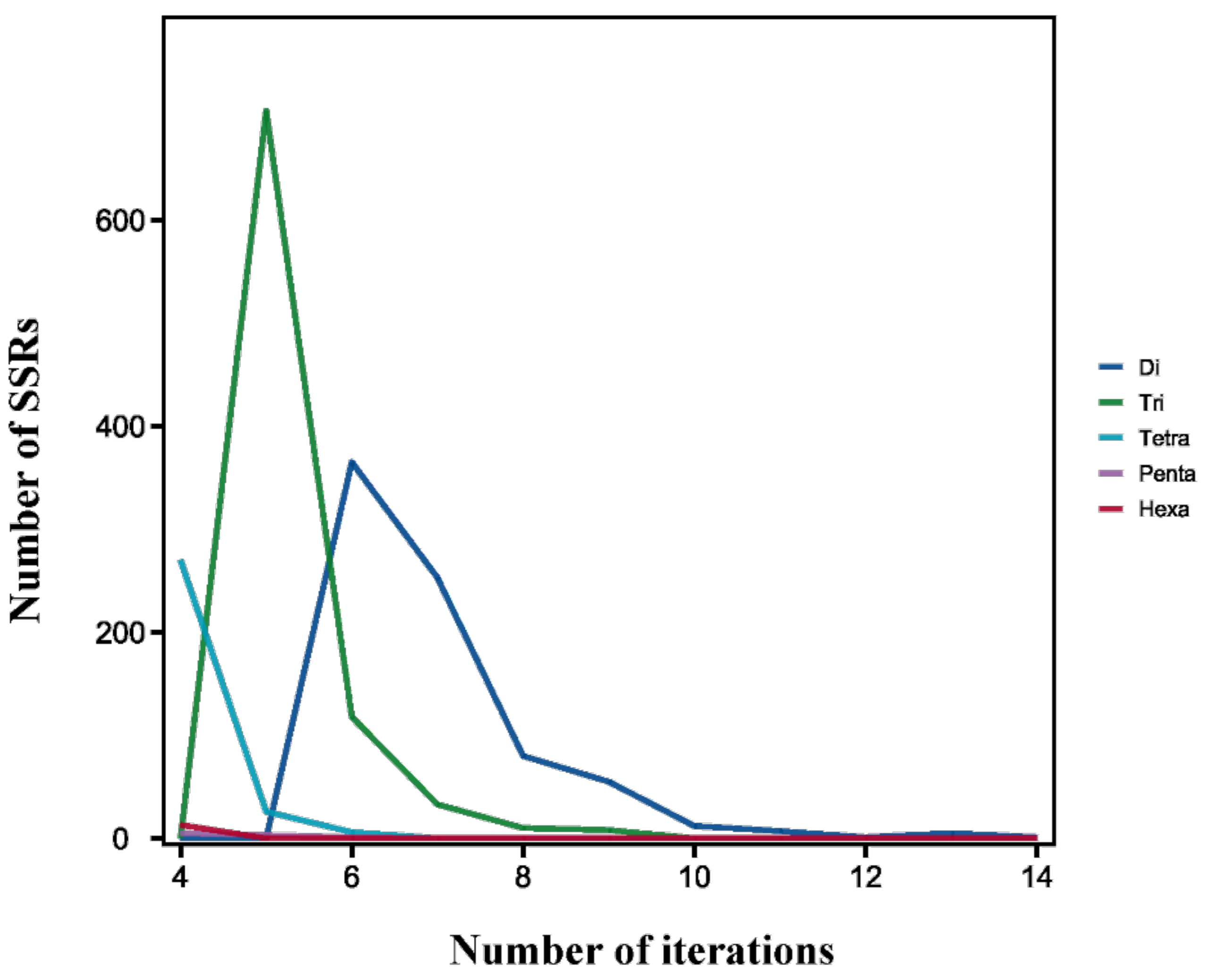
3.3. Predictive Analysis of SSRs
3.4. Verification of Novel and Polymorphic EST-SSRs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, H.; Zhang, D.J.; Xu, Y.J.; Wang, L.; Cheng, D.F.; Qi, Y.X.; Zeng, L.; Lu, Y.Y. Invasion, expansion, and control of Bactrocera dorsalis (Hendel) in China. J. Integr. Agric. 2019, 18, 771–787. [Google Scholar] [CrossRef]
- Aketarawong, N.; Bonizzoni, M.; Thanaphum, S.; Gomulski, L.M.; Gasperi, G.; Malacrida, A.R.; Gugliemino, C.R. Inferences on the population structure and colonization process of the invasive oriental fruit fly, Bactrocera dorsalis (Mendel). Mol. Ecol. 2007, 16, 3522–3532. [Google Scholar] [CrossRef]
- Clarke, A.R.; Armstrong, K.F.; Carmichael, A.E.; Milne, J.R.; Raghu, S.; Roderick, G.K.; Yeates, D.K. Invasive phytophagous pests arising through a recent tropical evolutionary radiation: The Bactrocera dorsalis complex of fruit flies. Annu. Rev. Entomol. 2005, 50, 293–319. [Google Scholar] [CrossRef] [Green Version]
- Stephens, A.E.A.; Kriticos, D.J.; Leriche, A. The current and future potential geographical distribution of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). Bull. Entomol. Res. 2007, 97, 369–378. [Google Scholar] [CrossRef]
- Shen, G.M.; Wang, X.N.; Dou, W.; Wang, J.J. Biochemical and molecular characterisation of acetylcholinesterase in four field populations of Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). Pest Manag. Sci. 2012, 68, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, E.; Gemmell, N.J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.K.; Chai, L.; Mayer, C.; Xu, Q.; Guo, W.; Deng, X. Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol. Biol. Rep. 2012, 39, 5373–5386. [Google Scholar] [CrossRef]
- Hina, F.; Yisilam, G.; Wang, S.Y.; Li, P.; Fu, C.X. De novo transcriptome assembly, gene annotation and SSR marker development in the moon seed genus Menispermum (Menispermaceae). Front. Genet. 2020, 11, 380. [Google Scholar] [CrossRef]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Duan, H.; Zhang, A.; Zhao, C.; Yu, Y.; Chu, D. Characterization and molecular marker screening of EST-SSRs and their polymorphism compared with Genomic-SSRs in Frankliniella occidentalis (Thysanoptera: Thripidae). Acta Entomol. Sin. 2012, 55, 634–640. [Google Scholar]
- Duan, H.S.; Yu, Y.; Zhang, A.S.; Guo, D.; Tao, Y.L.; Chu, D. Genetic diversity and inferences on potential source areas of adventive Frankliniella Occidentalis (Thysanoptera: Thripidae) in Shandong, China based on mitochondrial and microsatellite markers. Fla. Entomol. 2013, 96, 964–973. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, X.; Pang, B.; Tan, Y.; Chang, J.; Gao, L. High-throughput discovery of microsatellite markers in Galeruca daurica (Coleoptera: Chrysomelidae) from a transcriptome database. Chin. J. Appl. Entomol. 2016, 53, 1058–1064. [Google Scholar]
- Wu, Z.; Gao, P.; Wen, J. Characteristic analysis of microsatellite in Eucryptorrhynchus chinensis transcriptome. J. Environ. Entomol. 2016, 38, 979–983. [Google Scholar]
- Guo, Y.Q.; Zhang, J.Z.; Yang, M.L.; Yan, L.Z.; Zhu, K.Y.; Guo, Y.P.; Ma, E.B. Comparative analysis of cytochrome P450-like genes from Locusta migratoria manilensis: Expression profiling and response to insecticide exposure. Insect Sci. 2012, 19, 75–85. [Google Scholar] [CrossRef]
- Li, D.; Zhao, P. Development of microsatellite markers based on the transcriptome data of Sclomina erinacea (Heteroptera: Reduviidae). Acta Entomol. Sin. 2019, 62, 694–702. [Google Scholar]
- Yu, H.; Frommer, M.; Robson, M.K.; Meats, A.W.; Shearman, D.C.A.; Sved, J.A. Microsatellite analysis of the Queensland fruit fly Bactrocera tryoni (Diptera: Tephritidae) indicates spatial structuring: Implications for population control. Bull. Entomol. Res. 2001, 91, 139–147. [Google Scholar] [PubMed]
- Solano, P.; de La Rocque, S.; de Meeus, T.; Cuny, G.; Duvallet, G.; Cuisance, D. Microsatellite DNA markers reveal genetic differentiation among populations of Glossina palpalis gambiensis collected in the agro-pastoral zone of Sideradougou, Burkina Faso. Insect Mol. Biol. 2000, 9, 433–439. [Google Scholar] [CrossRef]
- Luna, C.; Bonizzoni, M.; Cheng, Q.Y.; Robinson, A.S.; Aksoy, S.; Zheng, L.B. Microsatellite polymorphism in tsetse flies (Diptera: Glossinidae). J. Med. Entomol. 2001, 38, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, Y.; He, X.; Mei, T.; Chen, B. Identification, characteristics and distribution of microsatellites in the whole genome of Anopheles sinensis (Diptera: Culicidae). Acta Entomol. Sin. 2016, 59, 1058–1068. [Google Scholar]
- Bonizzoni, M.; Malacrida, A.R.; Guglielmino, C.R.; Gomulski, L.M.; Gasperi, G.; Zheng, L. Microsatellite polymorphism in the Mediterranean fruit fly, Ceratitis capitata. Insect Mol. Biol. 2000, 9, 251–261. [Google Scholar] [CrossRef]
- Ravel, S.; Herve, J.P.; Diarrassouba, S.; Kone, A.; Cuny, G. Microsatellite markers for population genetic studies in Aedes aegypti (Diptera: Culicidae) from Cote d’Ivoire: Evidence for a microgeographic genetic differentiation of mosquitoes from Bouake. Acta Trop. 2002, 82, 39–49. [Google Scholar] [CrossRef]
- Ouyang, P.; Kang, D.; Mo, X.; Tian, E.; Hu, Y.; Huang, R. Development and characterization of high-throughput EST-based SSR markers for Pogostemon cablin using transcriptome sequencing. Molecules 2018, 23, 2014. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.A. Optimizing depth and type of high-throughput sequencing data for microsatellite discovery. Appl. Plant Sci. 2019, 7, e11298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Samuels, T.D.; Wu, Y.Q. Development of 1030 genomic SSR markers in switchgrass. Theor. Appl. Genet. 2011, 122, 677–686. [Google Scholar] [CrossRef]
- Contreras, J.L.; Knoppers, B.M. The Genomic Commons. Annu. Rev. Genom. Hum. Genet. 2018, 19, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K.; et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liang, S.; Duan, J.; Wang, J.; Chen, S.; Cheng, Z.; Zhang, Q.; Liang, X.; Li, Y. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genom. 2012, 13, 90. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Fan, B.; Cao, Z.; Su, Q.; Wang, Y.; Zhang, Z.; Wu, J.; Tian, J. A deep sequencing analysis of transcriptomes and the development of EST-SSR markers in mungbean (Vigna radiata). J. Genet. 2016, 95, 527–535. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.; Lu, X.; Luo, J.; Tang, F. SMRT sequencing of the full-length transcriptome of Odontotermes formosanus (Shiraki) under Serratia marcescens treatment. Sci. Rep. 2020, 10, 15909. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in Fungi revealed by Single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Zhang, Y.; Cui, H.H.; Liu, J.W.; Wu, Y.Q.; Cheng, Y.; Xu, H.X.; Huang, X.X.; Li, S.T.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35 (Suppl. 2), W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Fugenschuh, M.; Gera, R.; Tagarelli, A. ANGEL: A synthetic model for airline network generation emphasizing layers. IEEE Trans. Netw. Sci. Eng. 2020, 7, 1977–1987. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pages, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.C.; Zang, F.Q.; Xie, X.M.; Ma, Y.; Zheng, Y.Q.; Zang, D.K. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis. Sci. Rep. 2020, 10, 16249. [Google Scholar] [CrossRef]
- Meng, X.; Hu, J.; Li, Y.; Ouyang, G. Analysis of SSR loci in transcriptome database of Conopomorpha sinensis Bradley (Lepidoptera: Gracilariidae). J. Environ. Entomol. 2017, 39, 1219–1224. [Google Scholar]
- Yuan, Y.; Zhang, L.; Wu, G.; Zhu, J. High-throughput discovery microsatellites in Tomicus yunnanensis (Coleoptera: Scolytinae). J. Environ. Entomol. 2014, 36, 166–170. [Google Scholar]
- Beldade, P.; Rudd, S.; Gruber, J.D.; Long, A.D. A wing expressed sequence tag resource for Bicyclus anynana butterflies, an evo-devo model. BMC Genom. 2006, 7, 130. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Tao, Y.; Xue, H.; Yuan, M. Analysis of microsatellite loci in Plodia interpunctella based on transcriptome dataset. Plant Prot. 2017, 43, 43–48. [Google Scholar]
- Zhu, J.Y.; Li, Y.H.; Yang, S.; Li, Q.W. De novo assembly and characterization of the global transcriptome for Rhyacionia leptotubula using Illumina paired-end sequencing. PLoS ONE 2013, 8, e81096. [Google Scholar] [CrossRef]
- Li, L.T.; Zhu, Y.B.; Ma, J.F.; Li, Z.Y.; Dong, Z.P. An Analysis of the Athetis lepigone transcriptome from four developmental Stages. PLoS ONE 2013, 8, e73911. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Lu, C.; Lin, H.; Shi, Y.; He, H.; Zhang, F.; Liang, G. Characteristics of the SSR loci in the Anoplophora chinensis transcriptome. Chin. J. Appl. Entomol. 2019, 56, 1299–1308. [Google Scholar]
- Wei, D.D.; Shi, J.X.; Zhang, X.X.; Chen, S.C.; Wei, D.; Wang, J.J. Analysis of microsatellite loci from Bactrocera dorsalis based on transcriptome dataset. Ying Yong Sheng Tai Xue Bao 2014, 25, 1799–1805. [Google Scholar]
- Wang, C.; Du, L.; Li, P.; Yang, M.; Li, W.; Shen, Y.; Zhang, X.; Yue, B. Distribution patterns of microsatellites in the genome of the German cockroach (Blattella germanica). Acta Entomol. Sin. 2015, 58, 1037–1045. [Google Scholar]
- Zhang, F.J.; Guo, H.Y.; Zheng, H.J.; Zhou, T.; Zhou, Y.J.; Wang, S.Y.; Fang, R.X.; Qian, W.; Chen, X.Y. Massively parallel pyrosequencing-based transcriptome analyses of small brown planthopper (Laodelphax striatellus), a vector insect transmitting rice stripe virus (RSV). BMC Genom. 2010, 11, 303. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Huang, J.; Du, L.; Li, W.; Yue, B.; Zhang, X. Comparison of microsatellites between the genomes of Tetranychus urticae and Ixodes scapularis. Sichuan J. Zool. 2013, 32, 481–486. [Google Scholar]
- Cook, N.; Aziz, N.; Hedley, P.E.; Morris, J.; Milne, L.; Karley, A.J.; Hubbard, S.F.; Russell, J.R. Transcriptome sequencing of an ecologically important graminivorous sawfly: A resource for marker development. Conserv. Genet. Resour. 2011, 3, 789–795. [Google Scholar] [CrossRef]
- Liu, Y.-D.; Hou, M.-L. Analysis of microsatellite information in EST resource of Nilaparvata lugens (Homoptera: Delphacidae). Acta Entomol. Sin. 2010, 53, 239–247. [Google Scholar]
- Sun, J.T.; Zhang, Y.K.; Ge, C.; Hong, X.Y. Mining and characterization of sequence tagged microsatellites from the brown planthopper Nilaparvata lugens. J. Insect Sci. 2011, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Leng, C.; Li, Y.; Hu, D.; Wu, J.; Li, Y. Analysis of the larval midgut transcriptome and SSR markers in Grapholitha molesta (Lepidoptera: Tortricidae). Acta Entomol. Sin. 2018, 61, 1272–1283. [Google Scholar]
- Liu, Y.; Fu, K.; He, J.; Guo, W. Verification SSR primers by datamining genome SSR loci in Leptinotarsa decemlineata. J. Environ. Entomol. 2018, 40, 633–644. [Google Scholar]
- Duan, Y.; Wu, R.; Luo, L.; Wu, Y.; Jiang, Y.; Miao, J.; Gong, Z. Characterization of SSRs from the ESTs in the wheat midge, Sitodiplosis mosellana (Gehin) (Diptera: Cecidomyiidae). Acta Entomol. Sin. 2011, 54, 1147–1154. [Google Scholar]
- Luo, M.; Zhang, H.; Bin, S.; Lin, J. High-throughput discovery of SSR genetic markers in the mealybug, Phenacoccus solenopsis (Hemiptera: Pseudococcidae), from its transcriptome database. Acta Entomol. Sin. 2014, 57, 395–400. [Google Scholar]
- Jin, Y.; Cong, B.; Wang, L.; Zhang, H.; Dong, H. An analysis of the transcriptome of Epacromius coerulipes (Orthoptera: Acrididae). Acta Entomol. Sin. 2015, 58, 817–825. [Google Scholar]
- Li, C.C.; Li, G.Y.; Wang, Y.; Peng, Y. Development of SSR markers based on transcriptome sequences of the Wolf spider pardosa pseudoannulata (Araneae: Lycosidae) 1. Entomol. News 2020, 129, 6–15. [Google Scholar] [CrossRef]
- Zhou, Z.; Han, Y.; Zhen, Y.; Chai, J. Identification, characteristics and distribution of SSR loci in the transcriptome and genome skimming of Gampsocleis gratiosa (Orthoptera: Tettigoniidae). J. Environ. Entomol. 2019, 41, 799–807. [Google Scholar]
- Mi, Z.; Li, A.; Ruan, C.; Li, G.; Du, W.; Long, Y.; Zhu, Y. Searching and analysis of EST-SSR markers from linkage group 12 of the silkworm, Bombyx mori. Acta Entomol. Sin. 2011, 54, 1223–1230. [Google Scholar]
- Li, B.; Xia, Q.-Y.; Lu, C.; Zhou, Z.-Y. Analysis of microsatellites derived from bee ESTs. Acta Genet. Sin. 2004, 31, 1089–1094. [Google Scholar] [PubMed]
- Li, W.; Zhang, L.; Cheng, Y.; Luo, L.; Jiang, X. High-throughput discovery of microsatellite markers based on transcriptome sequencing in the oriental armyworm, Mythimna separata (Walker). Acta Phytophylacica Sin. 2017, 44, 377–384. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Location | Population Code | Number | Host Plants | Geo-Coordinates | Collection Date | |
|---|---|---|---|---|---|---|
| City/Province | Town | |||||
| Beihai | Tieshangang | BHTSG | 8 | Guava | N21°31′45″ E109°25′18″ | 2020.10.24 |
| Hepu | HPSK | 8 | Citrus Reiculata Blanco | N21°44′36″ E109°22′52″ | 2020.10.24 | |
| Weizhou island | WZD | 8 | Pawpaw | N21°01′39″ E109°05′24″ | 2021.6.9 | |
| Yulin | Yuzhouqu | YLYZQ | 8 | Guava | N22°41′35″ E110°07′43″ | 2020.10.29 |
| Luchuan | YLLC | 8 | Guava | N22°9′39″ E110°14′33″ | 2020.11.1 | |
| Rongxian | RXRX | 8 | Citrus Reiculata Blanco | N22°48′33″ E110°28′47″ | 2020.11.25 | |
| Chongzuo | Longzhou | CZLZ | 8 | Green date, Tangerine | N25°16′16″ E110°19′51″ | 2020.3.31 |
| Jiangzhouqu | CZJZQ | 6 | Citrus Reiculata Blanco | N22°39′34″ E107°38′21″ | 2020.10.31 | |
| Baise | Tianyang | BSTY | 6 | Mango | N23°44′08″ E106°54′56″ | 2020.10.31 |
| Guigang | Gangnanqu | GGGNQ | 8 | CitrusGonggan, sugar orange | N23°01′55″ E109°49′41″ | 2020.10.25 |
| Pingnan | GGPN | 8 | Citrus Reiculata Blanco | N23°25′17″ E110°32′11″ | 2020.11.25 | |
| Guilin | Yongfu | GLYF | 8 | Momordica Grosvenori | N24°59′31″ E109°59′54″ | 2020.11.1 |
| Gongcheng | GLGC | 8 | Gongcheng Persimmon | N24°43′41″ E110°52′31″ | 2020.11.2 | |
| Wuzhou | Mengshan | WZMS | 8 | Sugar Orange | N24°11′38″ E110°31′29″ | 2020.10.28 |
| Fangchenggang | Shangsi | FCGSS | 8 | Guava | N22°1′33″ E108°0′3″ | 2020.9.17 |
| Nanning | Longan | NNLA | 8 | Mango | N23°9′57″ E107°41′46″ | 2020.7.17 |
| Hechi | Duan | HCDA | 6 | Citrus Reiculata Blanco | N23°58′45″ E108°5′57″ | 2020.11.16 |
| Laibin | Xincheng | LBXC | 4 | Citrus Reiculata Blanco | N23°50′54″ E108°49′54″ | 2020.11.1 |
| Qinzhou | Qinnanqu | QZQN | 8 | Guava | N21°56′19″ E108°39′25″ | 2021.7.26 |
| Guangdong, Guangzhou | GZ | 8 | Uncertain | N23°9′29″ E113°21′28″ | 2020.10.17 | |
| Luoyang, Henan | HN | 8 | Peach | N34°39′55″ E112°24′55″ | 2021. 5.1 | |
| Baoshan, Yunnan | YN | 6 | Persimmon | N25°7′16″ E99°9′57″ | 2020.2.5 | |
| Categories | Sequencing | Amount |
|---|---|---|
| Subreads | Number of subreads | 40,319,890 |
| Number of bases | 40,685,843,094 | |
| Average length | 1009 | |
| N50 | 1659 | |
| Circular consensus sequence (CCS) | Number of sequences | 535,241 |
| Number of bases | 940,115,109 | |
| Mean length | 1756 | |
| Full-length non-concatemer (FLNC) | Number of sequences | 386,916 |
| Mean length | 1622 | |
| High-quality isoforms | Number of sequences | 22,780 (99.30%) |
| Mean length | 1783 | |
| N50 | 2256 | |
| Comparison of reference genomes | Total mapped | 17,459 (76.64%) |
| Multiple mapped | 362 (1.59%) | |
| Unique mapped | 17,097 (75.05%) | |
| Gene expression | All mapped genes | 4912 |
| All mapped isoforms | 12,274 | |
| Known isoforms | 5365 (43.71%) | |
| Novel isoforms | 226 (1.84%) | |
| New isoforms | 6683 (54.45%) |
| Genus | Name of Species | Number of Isoforms | Percentage (%) | |
|---|---|---|---|---|
| Individual | Generic Species | |||
| Bactrocera | B. dorsalis | 4663 | 41.82 | 44.88 |
| B. cucurbitae | 178 | 1.60 | ||
| B. latifrons | 98 | 0.88 | ||
| B. oleae | 65 | 0.58 | ||
| Ceratitis | C. capitata | 164 | 1.47 | 1.47 |
| Drosophila | D. melanogaster | 87 | 0.78 | 2.46 |
| D. grimshawi | 71 | 0.64 | ||
| D. persimilis | 58 | 0.52 | ||
| D. sechellia | 43 | 0.39 | ||
| D. busckii | 15 | 0.13 | ||
| Rhagoletis | R. zephyria | 26 | 0.23 | 0.23 |
| KEGG Category | Sub-Pathways | Genes |
|---|---|---|
| Metabolism (57.81%) | ||
| Metabolic pathways | 869 | |
| Biosynthesis of secondary metabolites | 367 | |
| Oxidative phosphorylation | 330 | |
| Biosynthesis of antibiotics | 317 | |
| Microbial metabolism in diverse environments | 309 | |
| Carbon metabolism | 278 | |
| Glycolysis/Gluconeogenesis | 246 | |
| Biosynthesis of amino acids | 200 | |
| Cellular processes (11.49%) | ||
| Phagosome | 159 | |
| Lysosome | 107 | |
| Endocytosis | 85 | |
| Peroxisome | 46 | |
| Focal adhesion | 17 | |
| Adherens junction | 17 | |
| Tight junction | 17 | |
| Regulation of actin cytoskeleton | 17 | |
| Quorum sensing | 8 | |
| Regulation of autophagy | 5 | |
| Gap junction | 3 | |
| Organismal systems (5.46%) | ||
| Phototransduction—fly | 82 | |
| Longevity regulating pathway—multiple species | 47 | |
| Dorso-ventral axis formation | 19 | |
| Platelet activation | 14 | |
| Leukocyte transendothelial migration | 14 | |
| Thyroid hormone signaling pathway | 14 | |
| Oxytocin signaling pathway | 14 | |
| Environmental information processing (8.71%) | ||
| ECM–receptor interaction | 61 | |
| Hippo signaling pathway—fly | 54 | |
| Wnt signaling pathway | 30 | |
| Two-component system | 28 | |
| FoxO signaling pathway | 22 | |
| ABC transporters | 17 | |
| Phosphatidylinositol signaling system | 16 | |
| TGF-beta signaling pathway | 16 | |
| Rap1 signaling pathway | 14 | |
| Hippo signaling pathway | 14 | |
| Genetic information processing (32.79%) | ||
| Ribosome | 280 | |
| RNA transport | 170 | |
| Protein processing in endoplasmic reticulum | 151 | |
| RNA degradation | 92 | |
| Proteasome | 76 | |
| Spliceosome | 74 | |
| Aminoacyl-tRNA biosynthesis | 62 | |
| Ubiquitin mediated proteolysis | 54 | |
| Protein export | 47 | |
| mRNA surveillance pathway | 40 |
| Repeat Type | Repeat Number | Total | Percentage (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11~14 | |||
| Dinucleotide | 365 | 253 | 80 | 55 | 12 | 14 | 779 | 39.38 | ||
| Trinucleotide | 706 | 118 | 33 | 10 | 8 | 875 | 44.24 | |||
| Tetranucleotide | 270 | 26 | 6 | 302 | 15.27 | |||||
| Pentanucleotide | 5 | 3 | 1 | 9 | 0.46 | |||||
| Hexanucleotide | 13 | 13 | 0.66 | |||||||
| Total | 288 | 735 | 490 | 286 | 90 | 63 | 12 | 14 | 1978 | 100.00 |
| Percentage (%) | 14.56 | 37.16 | 24.77 | 14.46 | 4.55 | 3.19 | 0.61 | 0.71 | ||
| Name | Motif Type | Primer Sequence | Tm (°C) | Size (bp) |
|---|---|---|---|---|
| BdSSR1 (Isoform 2924) | (TACA)4 | GGCAACCAATAGAACTGGGA | 53 | 280 |
| GTGCAAAAGTGTGTGCGTTT | ||||
| BdSSR2 (Isoform 3402) | (AAAC)4 | CGCGAATACTACGGACTTTAGG | 53 | 278 |
| CAACCTACCCACATCTACACACA | ||||
| BdSSR3 (Isoform 3866) | (TATG)4 | ATATCACCGCCGTAGCAAAC | 52 | 279 |
| TTGGCGTCAATCATAGCGTA | ||||
| BdSSR4 (Isoform 4371) | (ACAT)4 | TGCCATATGGTTGCATCAGT | 49 | 180 |
| GAAGCGCGAATGAACAAAAT | ||||
| BdSSR5 (Isoform 4367) | (CAG)5 | AAAGTAAATGTTGCGGTCGG | 51 | 258 |
| GTATAGCGCCGGTGATGAGT | ||||
| BdSSR6 (Isoform4777) | (ATAC)4 | AGCCCAGAAACTCACAGCAT | 50 | 197 |
| AACCGCAACAAAACAATTCC | ||||
| BdSSR7 (Isoform 4926) | (TA)7 | CGATAGCGCCCTATTTGTGT | 48 | 144 |
| CATTTGCGGTGCATTATTTG | ||||
| BdSSR8 (Isoform 5257) | (TAG)5 | TGTGACGGGTTGCTACCATA | 49 | 166 |
| CGCAAAAACAAGACCCAAAT | ||||
| BdSSR9 (Isoform 84) | (GGC6) | GCGACAAACAGTGCTTACGA | 53 | 233 |
| CCGCTGCTGTAAGAGGACTT | ||||
| BdSSR10 (Isoform 1104) | (AAC)6 | GCTTGTTGTTGTTGTGGTGG | 53 | 238 |
| ACGAAACGAGTGCGAAGAGT | ||||
| BdSSR11 (Isoform 1807) | (ATAC)5 | TTGAAACGCGTTGAAAAGTG | 50 | 269 |
| CGTTGCACTCAGGACTACGA | ||||
| BdSSR12 (Isoform 2327) | (TACA)4 | CATCGGGAAGTGCCAGTTAT | 52 | 264 |
| TGCCCAACATGTTATCTGGA | ||||
| BdSSR13 (Isoform 2402) | (ATTT)4 | GCTGGCCTACTCAGCGTATC | 53 | 217 |
| CTGCCCCGGTTAAAGTACAA | ||||
| BdSSR14 (Isoform 2859) | (ACAT)4 | GCGAAAGCGTAAAGGTGTGT | 47 | 132 |
| TTCAAAGTTAATGCGAAGCA | ||||
| BdSSR15 (Isoform2922) | (TACA)4 | GTGCAAAAGTGTGTGCGTTT | 51 | 155 |
| TCATCGGCCAATTCGAGTAT | ||||
| BdSSR16 (Isoform 2923) | (TACA)4 | GTGCAAAAGTGTGTGCGTTT | 51 | 155 |
| TCATCGGCCAATTCGAGTAT | ||||
| BdSSR17 (Isoform 2923) | (CAG)5 | GCAAGAAAAGCAGCAAAACC | 51 | 170 |
| GCTCGGCGAGTAACTCATTC | ||||
| BdSSR18 (Isoform 3194) | (CAAGAG)4 | GGCCAAACAGAATGAGGAAA | 51 | 200 |
| GCTACTACGCTTTCTTGCGG | ||||
| BdSSR19 (Isoform 3393) | (AGC)5 | CAATAGTGCGAGCAGTCGAA | 51 | 173 |
| GCAACGTTTCGTGATTCTCA | ||||
| BdSSR20 (Isoform 3695) | (GCTCCA)4 | TATACGGCTCCCTACATCGC | 54 | 181 |
| CACTTGGTGCAACCAGCTTA | ||||
| BdSSR21 (Isoform 3954) | (ACAT)4 | ACACACGAAGCGGAAGAGTT | 53 | 278 |
| CTGCCTCTCGTGTTTGCTTA | ||||
| BdSSR22 (Isoform 4461) | (GCT)6 | GTAATTGTGCCGTTCGAGGT | 53 | 217 |
| CCGGACTGCTATCCACATTT | ||||
| BdSSR23 (Isoform 4478) | (AAC)5 | GTCAGCTCTGGAGTCGGAAC | 55 | 249 |
| GGTGGTGTCTGTTGTCGTTG | ||||
| BdSSR24 (Isoform 4488) | (ATAC)5 | AGCAGCTGAAGAGGAAGTGC | 53 | 250 |
| TATGTAGAAACGGTTCGGGC | ||||
| BdSSR25 (Isoform 4513) | (ATAC)4 | GCGAAGCGGACAAAAGTTAG | 52 | 192 |
| TTTCTGCACTTCGCACTATCA | ||||
| BdSSR26 (Isoform 4606) | (CAG)5 | CAGCGAACAGGAGCACATTA | 50 | 236 |
| CGTATTGCATCATTTGTGGC | ||||
| BdSSR27 (Isoform 4611) | (CAG)5 | CAGCGAACAGGAGCACATTA | 50 | 236 |
| CGTATTGCATCATTTGTGGC | ||||
| BdSSR28 (Isoform 4614) | (CAG)5 | CAGCGAACAGGAGCACATTA | 50 | 236 |
| CGTATTGCATCATTTGTGGC | ||||
| BdSSR29 (Isoform 4621) | (AT)7 | TGTATGTACGCACACCAGCA | 51 | 114 |
| AACACAAATGCGGCTTCTTT | ||||
| BdSSR30 (Isoform 4621) | (CAG)5 | CAGCGAACAGGAGCACATTA | 50 | 236 |
| CGTATTGCATCATTTGTGGC | ||||
| BdSSR31 (Isoform 4654) | (TATG)4 | AGTTTTCGCTGCCGCTATTA | 52 | 214 |
| CGGCCATCTCGTAGGTATGT | ||||
| BdSSR32 (Isoform 4707) | (AC)6 | GCTAGTTTGACGATGAGGGC | 53 | 174 |
| CAGCACGTAATTTGCTGCAC | ||||
| BdSSR33 (Isoform 4731) | (ACAT)4 | TCCAACAGCAAATTCGACAA | 48 | 234 |
| TCTCATAAAAGCGCATACAAAAA | ||||
| BdSSR34 (Isoform 4932) | (CATA)4 | CAACGCTCACTCGCTCATTA | 49 | 190 |
| AATGTTCCGAATTTTCGTCG | ||||
| BdSSR35 (Isoform 4950) | (CAA)5 | GGTGCTGGTGGCAGTTTATT | 54 | 142 |
| TTGTTGTAGCGGTGGTGGTA | ||||
| BdSSR36 (Isoform 4980) | (CA)7 | TCCATGAGATCGAATGCAAA | 49 | 278 |
| CGATTCTAACTGCGAACGAA | ||||
| BdSSR37 (Isoform 4992) | (CAA) 5 | ACTCGCATTGAATGGACACA | 52 | 174 |
| AAATGATGCTGCTGCTGATG | ||||
| BdSSR38 (Isoform 5040) | (ATAC) 4 | GGATACTAGTGGTGGTCCGC | 54 | 175 |
| GCAGCTAGGATGCACAACAA | ||||
| BdSSR39 (Isoform 5126) | (TACA) 4 | ACAGCCGAGTTTGAGCTTGT | 50 | 245 |
| TTGCATGAAAAGCAAACACC | ||||
| BdSSR40 (Isoform 5749) | (GCT)5 | AAGACGAAGAAGATGCGGAA | 51 | 157 |
| AAGACGAAGAAGATGCGGAA | ||||
| BdSSR41 (Isoform 5805) | (AGC)5 | ACAGCAACAACAGCAACAGC | 55 | 225 |
| TGTGTGCTAGAAGACGCACC |
| Dominant Repeat Type | Order | Species | Frequency (%) | Omics Level | Reference |
|---|---|---|---|---|---|
| Mononucleotide | Hemiptera | Cimex lectularius | 18.68 | Transcriptome | Li et al., 2019 |
| Stephanitis nashi | 26.87 | Transcriptome | Xie et al., 2019 | ||
| Lepidoptera | Conopomorpha sinensis | 15.25 | Transcriptome | Meng et al., 2017 | |
| Mythimna separata | 11.51 | Transcriptome | Li et al., 2017 | ||
| Grapholitha molesta | 13.16 | Transcriptome | Leng et al., 2018 | ||
| Thysanoptera | Frankliniella occidentalis | 18.95 | Transcriptome | Duan et al., 2012 | |
| Coleoptera | Galeruca daurica | 5.36 | Transcriptome | Zhang et al., 2016 | |
| Tenebrio molitor | 1.67 | NA | Zhu et al., 2013 | ||
| Tribolium castaneum * | 10.87 | Transcriptome | Zhang et al., 2008 | ||
| Leptinotarsa decemlineata | NA | Genome | Liu et al., 2018 | ||
| Diptera | Sitodiplosis mosellana | 13.47 | Transcriptome | Duan et al., 2011 | |
| Anopheles sinensis | NA | Genome | Wang et al., 2016 | ||
| Homoptera | Phenacoccus solenopsis | 6.33 | Genome | Luo et al., 2014 | |
| Dinucleotide | Lepidoptera | Plodia interpunctell | 8.25 | Transcriptome | Tang et al., 2017 |
| Rhyacionia leptotubula | 3.09 | Transcriptome | Zhu et al., 2013 | ||
| Hemiptera | Arma chinensis | 7.6 | NA | Li et al., 2019 | |
| Orthoptera | Epacromius coerulipes | 44. 17 | Transcriptome | Jin et al., 2015 | |
| Gampsocleis gratiosa * | 18.64 | Transcriptome | Zhou et al., 2019 | ||
| Hymenoptera | Apis mellifera | 10.804 | Genome | Zhao et al., 2007 | |
| Trinucleotide | Coleoptera | Tomicus yunnanensis | 1.29 | Transcriptome | Yuan et al., 2014 |
| Anoplophora chinensis | 25.31 | Transcriptome | Han et al., 2019 | ||
| Eucryptorrhynchus chinensis | 10.36 | Transcriptome | Wu et al., 2016 | ||
| Lepidoptera | Dolerus aeneus | NA | NA | Cook et al., 2011 | |
| Athetis lepigone | 2.96 | NA | Li et al., 2013 | ||
| Mythimna separata | 1.93 | Transcriptome | Hu et al., 2015 | ||
| Plutella xylostella | 6.59 | Transcriptome | Ke et al., 2013 | ||
| Hemiptera | Nilaparvata lugens | NA | Transcriptome | Liu et al., 2010 | |
| Sclomina erinacea | 5.67 | NA | Li et al.,2019 | ||
| Homoptera | Laodelphax striatellus | NA | Transcriptome | Zhang et al., 2010 | |
| Orthoptera | Gampsocleis gratiosa * | 39.38 | Genome | Zhou et al., 2019 | |
| Diptera | Anopheles sinensis (2014) | NA | Transcriptome | Zhou et al., 2018 | |
| Bactrocera dorsalis | NA | Transcriptome | Wei et al., 2014 | ||
| Blattaria | Blattella germanica | NA | Genome | Wang et al.,2015 | |
| Tetranucleotide | Lepidoptera | Bombyx mori | 26.51 | Transcriptome | Mi et al., 2011 |
| Pentanucleotide | NA | NA | NA | NA | |
| Hexanucleotide | Coleoptera | Tribolium castaneum * | 13.65 | Genome | Zhang et al., 2008 |
| Hymenoptera | Bee (Apis) | 10.52 | Transcriptome | Li et al., 2004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, H.; Wang, X.; Zheng, X.; Lu, W.; Qin, F.; Chen, C. Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel). Insects 2021, 12, 938. https://doi.org/10.3390/insects12100938
Ouyang H, Wang X, Zheng X, Lu W, Qin F, Chen C. Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel). Insects. 2021; 12(10):938. https://doi.org/10.3390/insects12100938
Chicago/Turabian StyleOuyang, Huili, Xiaoyun Wang, Xialin Zheng, Wen Lu, Fengping Qin, and Chao Chen. 2021. "Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel)" Insects 12, no. 10: 938. https://doi.org/10.3390/insects12100938
APA StyleOuyang, H., Wang, X., Zheng, X., Lu, W., Qin, F., & Chen, C. (2021). Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel). Insects, 12(10), 938. https://doi.org/10.3390/insects12100938