
Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome


Abstract
:Simple Summary
Abstract
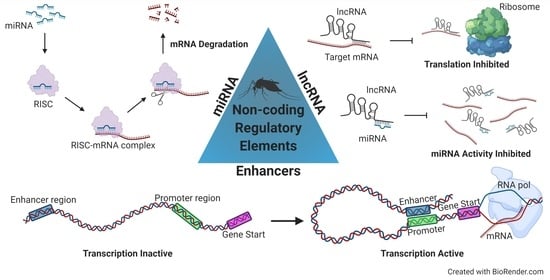
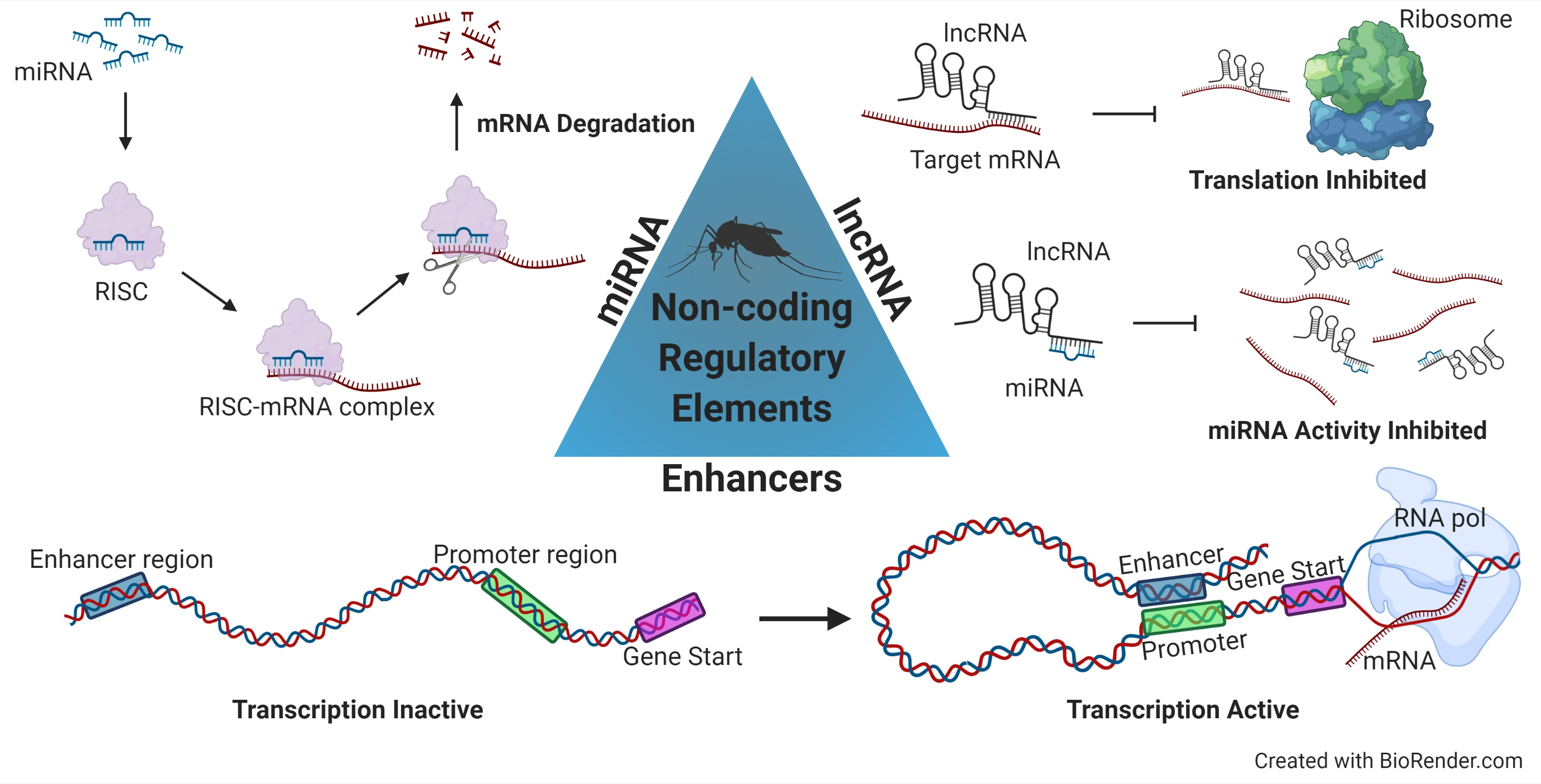
1. Introduction
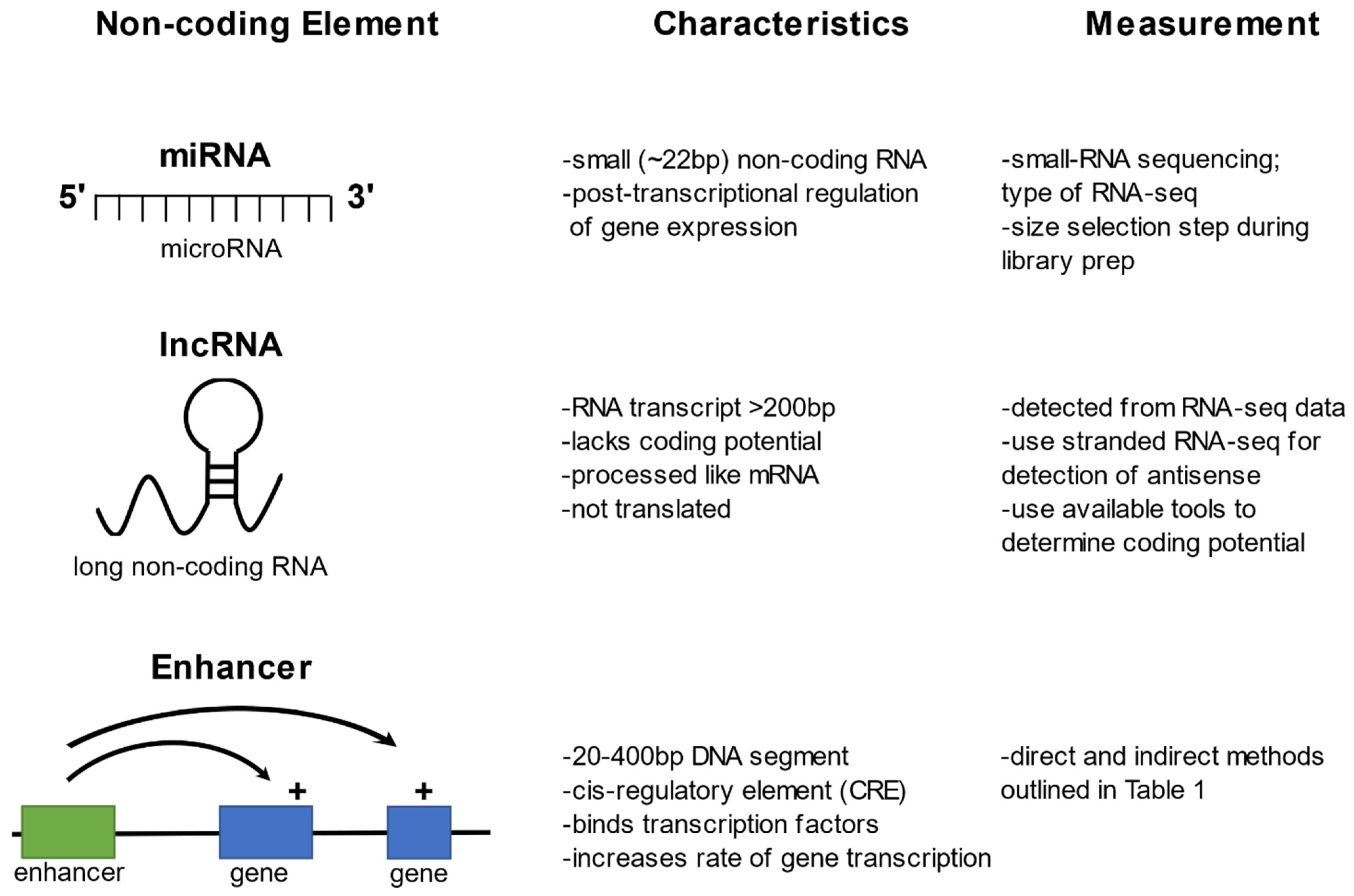
2. MicroRNAs
3. Long Non-Coding RNAs
4. Enhancers
Screening for Enhancers
5. Direct Methods for Enhancer Discovery
STARR-Seq
6. Indirect Methods for Enhancer Discovery
6.1. ATAC-Seq (Single Cell Capable)
6.2. ChIP-Seq
6.3. DNase-Seq (Single Cell Capable)
6.4. FAIRE-Seq
6.5. MNase-Seq (Single Cell Capable)
6.6. NOMe-Seq
7. Enhancer RNAs
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ohno, S. So much “junk” DNA in our genome. In Evolution of Genetic Systems; Smith, H.H., Ed.; Gordon and Breach: New York, NY, USA, 1972; pp. 366–370. [Google Scholar]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- The International HapMap 3 Consortium; Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Dermitzakis, E.; Schaffner, S.F.; Yu, F.; et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ayala, D.; Zhang, S.; Chateau, M.; Fouet, C.; Morlais, I.; Costantini, C.; Hahn, M.W.; Besansky, N. Association mapping desiccation resistance within chromosomal inversions in the African malaria vector Anopheles gambiae. Mol. Ecol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolliopoulou, A.; Santos, D.; Taning, C.N.T.; Wynant, N.; Vanden Broeck, J.; Smagghe, G.; Swevers, L. PIWI pathway against viruses in insects. Wiley Interdiscip Rev. RNA 2019, 10, e1555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.Y.; Palli, S.R. Mechanisms, Applications, and Challenges of Insect RNA Interference. Annu. Rev. Entomol. 2020, 65, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Biggar, K.K.; Storey, K.B. Functional impact of microRNA regulation in models of extreme stress adaptation. J. Mol. Cell Biol. 2018, 10, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhou, S.; Wang, J.; Hu, W. microRNA profiles and functions in mosquitoes. PLoS Negl. Trop. Dis. 2018, 12, e0006463. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Miesen, P.; van Rij, R.P. Antiviral RNAi in Insects and Mammals: Parallels and Differences. Viruses 2019, 11, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutvágner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.; Tuschl, T. RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 2004, 18, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bhartiya, D.; Scaria, V. Genomic variations in non-coding RNAs: Structure, function and regulation. Genomics 2016, 107, 59–68. [Google Scholar] [CrossRef]
- Bensen, J.T.; Graff, M.; Young, K.L.; Sethupathy, P.; Parker, J.; Pecot, C.V.; Currin, K.; Haddad, S.A.; Ruiz-Narvaez, E.A.; Haiman, C.A.; et al. A survey of microRNA single nucleotide polymorphisms identifies novel breast cancer susceptibility loci in a case-control, population-based study of African-American women. Breast Cancer Res. 2018, 20, 45. [Google Scholar] [CrossRef]
- Carissimo, G.; Pain, A.; Belda, E.; Vernick, K.D. Highly focused transcriptional response of Anopheles coluzzii to O’nyong nyong arbovirus during the primary midgut infection. BMC Genom. 2018, 19, 526. [Google Scholar] [CrossRef] [Green Version]
- Lampe, L.; Levashina, E.A. The role of microRNAs in Anopheles biology-an emerging research field. Parasite Immunol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Dennison, N.J.; BenMarzouk-Hidalgo, O.J.; Dimopoulos, G. MicroRNA-regulation of Anopheles gambiae immunity to Plasmodium falciparum infection and midgut microbiota. Dev. Comp. Immunol. 2015, 49, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.; Kokoza, V.A.; Zhang, C.; Aksoy, E.; Raikhel, A.S. MicroRNA-277 targets insulin-like peptides 7 and 8 to control lipid metabolism and reproduction in Aedes aegypti mosquitoes. Proc. Natl. Acad. Sci. USA 2017, 114, E8017–E8024. [Google Scholar] [CrossRef] [Green Version]
- Git, A.; Dvinge, H.; Salmon-Divon, M.; Osborne, M.; Kutter, C.; Hadfield, J.; Bertone, P.; Caldas, C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010, 16, 991–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Friedländer, M.R. Computational Prediction of miRNA Genes from Small RNA Sequencing Data. Front. Bioeng. Biotechnol. 2015, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Haac, M.E.; Anderson, M.A.; Eggleston, H.; Myles, K.M.; Adelman, Z.N. The hub protein loquacious connects the microRNA and short interfering RNA pathways in mosquitoes. Nucleic Acids Res. 2015, 43, 3688–3700. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hao, Z.; Huang, L.; Chen, L.; Wei, Q.; Cai, L.; Liang, S. Comparative expression profile of microRNAs in Anopheles anthropophagus midgut after blood-feeding and Plasmodium infection. Parasites Vectors 2017, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raabe, C.A.; Tang, T.H.; Brosius, J.; Rozhdestvensky, T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014, 42, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Witwer, K.W.; Halushka, M.K. Toward the promise of microRNAs-Enhancing reproducibility and rigor in microRNA research. RNA Biol. 2016, 13, 1103–1116. [Google Scholar] [CrossRef]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [Green Version]
- Samuel, G.H.; Adelman, Z.N.; Myles, K.M. Antiviral Immunity and Virus-Mediated Antagonism in Disease Vector Mosquitoes. Trends Microbiol. 2018, 26, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mead, E.A.; Liang, S.; Tu, Z. Direct sequencing and expression analysis of a large number of miRNAs in Aedes aegypti and a multi-species survey of novel mosquito miRNAs. BMC Genom. 2009, 10, 581. [Google Scholar] [CrossRef] [Green Version]
- Osei-Amo, S.; Hussain, M.; O’Neill, S.L.; Asgari, S. Wolbachia-induced aae-miR-12 miRNA negatively regulates the expression of MCT1 and MCM6 genes in Wolbachia-infected mosquito cell line. PLoS ONE 2012, 7, e50049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Rana, V.; Shrinet, J.; Sharma, A.; Tridibes, A.; Sunil, S.; Bhatnagar, R.K. Blood feeding and Plasmodium infection alters the miRNome of Anopheles stephensi. PLoS ONE 2014, 9, e98402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila-Bonilla, R.G.; Yocupicio-Monroy, M.; Marchat, L.A.; Pérez-Ishiwara, D.G.; Cerecedo-Mercado, D.A.; Del Ángel, R.M.; Salas-Benito, J.S. miR-927 has pro-viral effects during acute and persistent infection with dengue virus type 2 in C6/36 mosquito cells. J. Gen. Virol. 2020, 101, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wang, G.; Li, C.; Xing, D.; Yan, T.; Zhu, X.; Liu, Q.; Wu, Q.; Guo, X.; Zhao, T. Screening for differentially expressed miRNAs in Aedes albopictus (Diptera: Culicidae) exposed to DENV-2 and their effect on replication of DENV-2 in C6/36 cells. Parasit Vectors 2019, 12, 44. [Google Scholar] [CrossRef]
- Dubey, S.K.; Shrinet, J.; Sunil, S. Aedes aegypti microRNA, miR-2944b-5p interacts with 3’UTR of chikungunya virus and cellular target vps-13 to regulate viral replication. PLoS Negl. Trop. Dis. 2019, 13, e0007429. [Google Scholar] [CrossRef]
- Trobaugh, D.W.; Sun, C.; Bhalla, N.; Gardner, C.L.; Dunn, M.D.; Klimstra, W.B. Cooperativity between the 3’ untranslated region microRNA binding sites is critical for the virulence of eastern equine encephalitis virus. PLoS Pathog. 2019, 15, e1007867. [Google Scholar] [CrossRef] [Green Version]
- Yen, P.S.; Chen, C.H.; Sreenu, V.; Kohl, A.; Failloux, A.B. Assessing the Potential Interactions between Cellular miRNA and Arboviral Genomic RNA in the Yellow Fever Mosquito, Aedes aegypti. Viruses 2019, 11, 540. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.B.; Asgari, S. Ross River Virus Provokes Differentially Expressed MicroRNA and RNA Interference Responses in Aedes aegypti Mosquitoes. Viruses 2020, 12, 695. [Google Scholar] [CrossRef]
- Lampe, L.; Jentzsch, M.; Kierszniowska, S.; Levashina, E.A. Metabolic balancing by miR-276 shapes the mosquito reproductive cycle and Plasmodium falciparum development. Nat. Commun. 2019, 10, 5634. [Google Scholar] [CrossRef] [Green Version]
- Lampe, L.; Levashina, E.A. MicroRNA Tissue Atlas of the Malaria Mosquito Anopheles gambiae. G3 (Bethesda) 2018, 8, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Meuti, M.E.; Bautista-Jimenez, R.; Reynolds, J.A. Evidence that microRNAs are part of the molecular toolkit regulating adult reproductive diapause in the mosquito, Culex pipiens. PLoS ONE 2018, 13, e0203015. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Scheel, T.K.; Luna, J.M.; Park, C.Y.; Fak, J.J.; Nishiuchi, E.; Rice, C.M.; Darnell, R.B. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 2015, 6, 8864. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Fu, X.; Dong, Y.; Simões, M.L.; Zhu, J.; Dimopoulos, G. Broad spectrum immunomodulatory effects of Anopheles gambiae microRNAs and their use for transgenic suppression of Plasmodium. PLoS Pathog. 2020, 16, e1008453. [Google Scholar] [CrossRef]
- Fu, X.; Liu, P.; Dimopoulos, G.; Zhu, J. Dynamic miRNA-mRNA interactions coordinate gene expression in adult Anopheles gambiae. PLOS Genet. 2020, 16, e1008765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, B.; Roy, S.; Saha, T.T.; Kokoza, V.A.; Li, M.; Raikhel, A.S. microRNA-309 targets the Homeobox gene SIX4 and controls ovarian development in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. USA 2016, 113, E4828–E4836. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonasio, R.; Shiekhattar, R. Regulation of transcription by long noncoding RNAs. Annu. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardini, A.; Shiekhattar, R. The many faces of long noncoding RNAs. FEBS J. 2015, 282, 1647–1657. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Militello, G.; Weirick, T.; John, D.; Doring, C.; Dimmeler, S.; Uchida, S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform. 2017, 18, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Shastry, B.S. SNPs: Impact on gene function and phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar] [CrossRef]
- Legeai, F.; Derrien, T. Identification of long non-coding RNAs in insects genomes. Curr. Opin. Insect Sci. 2015, 7, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Maciel, L.F.; Morales-Vicente, D.A.; Silveira, G.O.; Ribeiro, R.O.; Olberg, G.G.O.; Pires, D.S.; Amaral, M.S.; Verjovski-Almeida, S. Weighted Gene Co-Expression Analyses Point to Long Non-Coding RNA Hub Genes at Different Schistosoma mansoni Life-Cycle Stages. Front. Genet. 2019, 10, 823. [Google Scholar] [CrossRef]
- Jenkins, A.M.; Waterhouse, R.M.; Muskavitch, M.A. Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genom. 2015, 16, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padron, A.; Molina-Cruz, A.; Quinones, M.; Ribeiro, J.M.; Ramphul, U.; Rodrigues, J.; Shen, K.; Haile, A.; Ramirez, J.L.; Barillas-Mury, C. In depth annotation of the Anopheles gambiae mosquito midgut transcriptome. BMC Genom. 2014, 15, 636. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [Green Version]
- Mei-zhen, L.; Hua-mei, X.; Kang, H.; Fei, L. Progress and prospects of noncoding RNAs in insects. J. Integr. Agric. 2019, 18, 729–747. [Google Scholar]
- Wang, Y.; Xu, T.; He, W.; Shen, X.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics 2018, 110, 35–42. [Google Scholar] [CrossRef]
- Etebari, K.; Furlong, M.J.; Asgari, S. Genome wide discovery of long intergenic non-coding RNAs in Diamondback moth (Plutella xylostella) and their expression in insecticide resistant strains. Sci. Rep. 2015, 5, 14642. [Google Scholar] [CrossRef]
- Chen, M.J.; Chen, L.K.; Lai, Y.S.; Lin, Y.Y.; Wu, D.C.; Tung, Y.A.; Liu, K.Y.; Shih, H.T.; Chen, Y.J.; Lin, Y.L.; et al. Integrating RNA-seq and ChIP-seq data to characterize long non-coding RNAs in Drosophila melanogaster. BMC Genom. 2016, 17, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.S.; Marques, A.C.; Tibbit, C.; Haerty, W.; Bassett, A.R.; Liu, J.L.; Ponting, C.P. Identification and properties of 1119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol. Evol. 2012, 4, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Wu, J.; Zhou, S.; Wang, J.; Hu, W. Characterization and potential role of microRNA in the Chinese dominant malaria mosquito Anopheles sinensis (Diptera: Culicidae) throughout four different life stages. Cell Biosci. 2018, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Kato, Y.; Perez, C.A.G.; Mohamad Ishak, N.S.; Nong, Q.D.; Sudo, Y.; Matsuura, T.; Wada, T.; Watanabe, H. A 5′ UTR-Overlapping LncRNA Activates the Male-Determining Gene doublesex1 in the Crustacean Daphnia magna. Curr. Biol. 2018, 28, 1811–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic Identification and Characterization of Long Non-Coding RNAs in the Silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, B.B.; Olcese, U.; Cabrera, J.R.; Horabin, J.I. An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision. Biochim. Biophys. Acta 2014, 1839, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Giraldo-Calderon, G.I.; Emrich, S.J.; MacCallum, R.M.; Maslen, G.; Dialynas, E.; Topalis, P.; Ho, N.; Gesing, S.; VectorBase, C.; Madey, G.; et al. VectorBase: An updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2015, 43, D707–D713. [Google Scholar] [CrossRef]
- Matthews, B.J.; Dudchenko, O.; Kingan, S.B.; Koren, S.; Antoshechkin, I.; Crawford, J.E.; Glassford, W.J.; Herre, M.; Redmond, S.N.; Rose, N.H.; et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 2018, 563, 501–507. [Google Scholar] [CrossRef]
- Etebari, K.; Asad, S.; Zhang, G.; Asgari, S. Identification of Aedes aegypti Long Intergenic Non-coding RNAs and Their Association with Wolbachia and Dengue Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0005069. [Google Scholar] [CrossRef]
- Etebari, K.; Hegde, S.; Saldana, M.A.; Widen, S.G.; Wood, T.G.; Asgari, S.; Hughes, G.L. Global Transcriptome Analysis of Aedes aegypti Mosquitoes in Response to Zika Virus Infection. mSphere 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Azlan, A.; Obeidat, S.M.; Yunus, M.A.; Azzam, G. Systematic identification and characterization of Aedes aegypti long noncoding RNAs (lncRNAs). Sci. Rep. 2019, 9, 12147. [Google Scholar] [CrossRef] [PubMed]
- Azlan, A.; Halim, M.A.; Mohamad, F.; Azzam, G. Identification and characterization of long noncoding RNAs and their association with acquisition of blood meal in Culex quinquefasciatus. Insect Sci. 2020. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, Y.; Xu, Y.; Lai, Z.; Jin, B.; Hao, Y.; Gao, Y.; Sun, Y.; Chen, X.G.; Gu, J. Differentiation of Long Non-Coding RNA and mRNA Expression Profiles in Male and Female Aedes albopictus. Front. Genet. 2019, 10, 975. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef]
- Catarino, R.R.; Neumayr, C.; Stark, A. Promoting transcription over long distances. Nat. Genet. 2017, 49, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 2011, 471, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanoski, C.E.; Link, V.M.; Heinz, S.; Glass, C.K. Exploiting genomics and natural genetic variation to decode macrophage enhancers. Trends Immunol. 2015, 36, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Sicard, A.; Kappel, C.; Lee, Y.W.; Wozniak, N.J.; Marona, C.; Stinchcombe, J.R.; Wright, S.I.; Lenhard, M. Standing genetic variation in a tissue-specific enhancer underlies selfing-syndrome evolution in Capsella. Proc. Natl. Acad. Sci. USA 2016, 113, 13911–13916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017, 49, 1602–1612. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Spies, D.; Matts, J.A.; Sytnikova, Y.A.; Pagani, M.; Lau, N.C.; Stark, A. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat. Genet. 2014, 46, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchini, L.F.; Pollard, K.S. Can a few non-coding mutations make a human brain? Bioessays 2015, 37, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Vierstra, J.; Rynes, E.; Sandstrom, R.; Zhang, M.; Canfield, T.; Hansen, R.S.; Stehling-Sun, S.; Sabo, P.J.; Byron, R.; Humbert, R.; et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 2014, 346, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Tomoyasu, Y.; Halfon, M.S. How to study enhancers in non-traditional insect models. J. Exp. Biol. 2020, 223 (Suppl. S1). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezcano, O.M.; Sánchez-Polo, M.; Ruiz, J.L.; Gómez-Díaz, E. Chromatin Structure and Function in Mosquitoes. Front. Genet. 2020. [Google Scholar] [CrossRef]
- Wold, B.; Myers, R.M. Sequence census methods for functional genomics. Nat. Methods 2008, 5, 19–21. [Google Scholar] [CrossRef]
- Gould, S.J.; Subramani, S. Firefly luciferase as a tool in molecular and cell biology. Anal. Biochem. 1988, 175, 5–13. [Google Scholar] [CrossRef]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- O’Brochta, D.A.; Pilitt, K.L.; Harrell, R.A.; Aluvihare, C., 2nd; Alford, R.T. Gal4-based enhancer-trapping in the malaria mosquito Anopheles stephensi. G3 (Bethesda) 2012, 2, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, F.; Ahituv, N. Decoding enhancers using massively parallel reporter assays. Genomics 2015, 106, 159–164. [Google Scholar] [CrossRef]
- Muerdter, F.; Boryn, L.M.; Arnold, C.D. STARR-seq-principles and applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumayr, C.; Pagani, M.; Stark, A.; Arnold, C.D. STARR-seq and UMI-STARR-seq: Assessing Enhancer Activities for Genome-Wide-, High-, and Low-Complexity Candidate Libraries. Curr. Protoc Mol. Biol. 2019, 128, e105. [Google Scholar] [CrossRef] [Green Version]
- Buerger, A. BasicSTARRseq: Basic Peak Calling on STARR-Seq Data. R Package Version 1.18.0. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/BasicSTARRseq.html (accessed on 22 February 2021).
- Lee, D.; Shi, M.; Moran, J.; Wall, M.; Zhang, J.; Liu, J.; Fitzgerald, D.; Kyono, Y.; Ma, L.; White, K.P.; et al. STARRPeaker: Uniform processing and accurate identification of STARR-seq active regions. Genome Biol. 2020, 21, 298. [Google Scholar] [CrossRef]
- Nardini, L.; Holm, I.; Pain, A.; Bischoff, E.; Gohl, D.M.; Zongo, S.; Guelbeogo, W.M.; Sagnon, N.; Vernick, K.D.; Riehle, M.M. Influence of genetic polymorphism on transcriptional enhancer activity in the malaria vector Anopheles coluzzii. Sci. Rep. 2019, 9, 15275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109. [Google Scholar] [CrossRef]
- Shashikant, T.; Ettensohn, C.A. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019, 151, 219–235. [Google Scholar] [CrossRef]
- Baek, S.; Lee, I. Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 2020, 18, 1429–1439. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Zhang, G.; Esteve, P.O.; Spracklin, G.; Sian, S.; Xu, S.Y.; Benoukraf, T.; Pradhan, S. NicE-seq: High resolution open chromatin profiling. Genome Biol. 2017, 18, 122. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.L.; Ranford-Cartwright, L.C.; Gomez-Diaz, E. The regulatory genome of the malaria vector Anopheles gambiae: Integrating chromatin accessibility and gene expression. bioRxiv 2020. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Siegel, T.N.; Hekstra, D.R.; Kemp, L.E.; Figueiredo, L.M.; Lowell, J.E.; Fenyo, D.; Wang, X.; Dewell, S.; Cross, G.A. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009, 23, 1063–1076. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, J.L.; Yerbanga, R.S.; Lefevre, T.; Ouedraogo, J.B.; Corces, V.G.; Gomez-Diaz, E. Chromatin changes in Anopheles gambiae induced by Plasmodium falciparum infection. Epigenet. Chromatin 2019, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Diaz, E.; Rivero, A.; Chandre, F.; Corces, V.G. Insights into the epigenomic landscape of the human malaria vector Anopheles gambiae. Front. Genet. 2014, 5, 277. [Google Scholar] [CrossRef] [Green Version]
- Lukyanchikova, V.; Nuriddinov, M.; Belokopytova, P.; Liang, J.; Reijnders, M.J.M.F.; Ruzzante, L.; Waterhouse, R.M.; Tu, Z.; Sharakhov, I.V.; Fishman, V. Anopheles mosquitoes revealed new principles of 3D genome organization in insects. bioRxiv 2020, 2020, 114017. [Google Scholar] [CrossRef]
- Sim, C.; Kang, D.S.; Kim, S.; Bai, X.; Denlinger, D.L. Identification of FOXO targets that generate diverse features of the diapause phenotype in the mosquito Culex pipiens. Proc. Natl. Acad. Sci. USA 2015, 112, 3811–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Galas, D.J.; Schmitz, A. DNAse footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978, 5, 3157–3170. [Google Scholar] [CrossRef]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenet. Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, D.Q.; Shaffer, J.J.; Chavez, C.A.; Douglass, P.L. Identification and mapping of the promoter for the gene encoding the ferritin heavy-chain homologue of the yellow fever mosquito Aedes aegypti. Insect Biochem. Mol. Biol. 2003, 33, 51–62. [Google Scholar] [CrossRef]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giresi, P.G.; Lieb, J.D. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 2009, 48, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Behura, S.K.; Sarro, J.; Li, P.; Mysore, K.; Severson, D.W.; Emrich, S.J.; Duman-Scheel, M. High-throughput cis-regulatory element discovery in the vector mosquito Aedes aegypti. BMC Genom. 2016, 17, 341. [Google Scholar] [CrossRef] [Green Version]
- Perez-Zamorano, B.; Rosas-Madrigal, S.; Lozano, O.A.M.; Castillo Mendez, M.; Valverde-Garduno, V. Identification of cis-regulatory sequences reveals potential participation of lola and Deaf1 transcription factors in Anopheles gambiae innate immune response. PLoS ONE 2017, 12, e0186435. [Google Scholar] [CrossRef] [PubMed]
- Axel, R. Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry 1975, 14, 2921–2925. [Google Scholar] [CrossRef] [PubMed]
- Kuan, P.F.; Huebert, D.; Gasch, A.; Keles, S. A non-homogeneous hidden-state model on first order differences for automatic detection of nucleosome positions. Stat. Appl. Genet. Mol. Biol. 2009, 8, 29. [Google Scholar] [CrossRef]
- Mieczkowski, J.; Cook, A.; Bowman, S.K.; Mueller, B.; Alver, B.H.; Kundu, S.; Deaton, A.M.; Urban, J.A.; Larschan, E.; Park, P.J.; et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016, 7, 11485. [Google Scholar] [CrossRef]
- Kensche, P.R.; Hoeijmakers, W.A.; Toenhake, C.G.; Bras, M.; Chappell, L.; Berriman, M.; Bartfai, R. The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 2016, 44, 2110–2124. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Vera, D.L.; Hughes, K.A.; Dennis, J.H. Stimulation of the Drosophila immune system alters genome-wide nucleosome occupancy. Genom. Data 2015, 3, 146–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lay, F.D.; Kelly, T.K.; Jones, P.A. Nucleosome Occupancy and Methylome Sequencing (NOMe-seq). Methods Mol. Biol. 2018, 1708, 267–284. [Google Scholar] [CrossRef]
- Rhie, S.K.; Schreiner, S.; Farnham, P.J. Defining Regulatory Elements in the Human Genome Using Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq). Methods Mol. Biol. 2018, 1766, 209–229. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [Green Version]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaylichenko, O.; Bondarenko, V.; Harnett, D.; Schor, I.E.; Males, M.; Viales, R.R.; Furlong, E.E.M. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 2018, 32, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Small, S.; Arnosti, D.N. Transcriptional Enhancers in Drosophila. Genetics 2020, 216, 1–26. [Google Scholar] [CrossRef]
- The Anopheles Gambiae 1000 Genomes Consortium; Data Analysis Group; Miles, A. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 2017, 552, 96–100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Method | Date of First Publication a | Protocol | Time Needed c | Number of Mosquitoes Needed d | Previous Use in Mosquito Disease Vectors | Protocol Bias |
|---|---|---|---|---|---|---|
| ATAC-seq Insert known sequence tags into open DNA | 2013 [117] (543 ref) | Tagmentation DNA purification DNA labeling Sequencing |  |  | Aedes aegypti: [80] Anopheles gambiae: [116] | Generates non-specific amplification of extra-nuclear DNA (mitochondrial)[114] |
| CHIP-seq Immunoprecipitate open DNA | 2007 [140] (4382 ref) | Crosslink proteins to DNA Shear DNA Immunoprecipitation of open DNA Sequencing |  |  | Anopheles atroparvus: [122] Culex pipiens: [123] Anopheles gambiae: [120,121] | Antibody availability and specificity [114] |
| DNase-seq Enzymatically remove open DNA | 2008 [124] (194 ref) | DNaseI DNA digestion Gel electrophoresis Sequencing | | ND | None | Dnase I cleavage bias [127] |
| FAIRE-seq Crosslinking and extracting open DNA | 2009 [130] (60 ref) | DNA crosslinked and sheared Phenol-Chloroform extraction Sequencing |  |  | Aedes aegypti: [131] Anopheles gambiae: [132] | Low signal to noise ratio, variation in formaldehyde fixation step [126] |
| MNase-seq Enzymatically remove nucleosomal DNA | 2009 [134] (90 ref) | Mnase DNA digestion Nucleosomal DNA purified Sequencing | | ND | None | Variable Mnase digestion [126] |
| NOMe-seq Methylate accessible DNA | 2011 [141] (19 ref) | Cells fixed and sheared M.CvPi b methylation of GC dinucleotides Bisulfite Conversion Sequencing | | ND | None | Requires specific library fragment size to minimize bias towards CpG islands [139] |
| STARR-seq Quantitatively assesses enhancer activity of genomic fragments on a genome-wide scale | 2013 [102] (27 ref) | Genomic DNA fragmented Addition of linkers Cloned into vector Cell transfection mRNA isolation and cDNA generation Sequencing | | | None | Does not capture conditional states, catalogs all enhancers [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farley, E.J.; Eggleston, H.; Riehle, M.M. Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome. Insects 2021, 12, 186. https://doi.org/10.3390/insects12020186
Farley EJ, Eggleston H, Riehle MM. Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome. Insects. 2021; 12(2):186. https://doi.org/10.3390/insects12020186
Chicago/Turabian StyleFarley, Elise J., Heather Eggleston, and Michelle M. Riehle. 2021. "Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome" Insects 12, no. 2: 186. https://doi.org/10.3390/insects12020186
APA StyleFarley, E. J., Eggleston, H., & Riehle, M. M. (2021). Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome. Insects, 12(2), 186. https://doi.org/10.3390/insects12020186