
Three Complete Mitochondrial Genomes of Erotylidae (Coleoptera: Cucujoidea) with Higher Phylogenetic Analysis


 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Genomic DNA Extraction
2.2. Genome Sequencing and Analyses
2.3. Phylogenetic Analyses
3. Results and Discussion
3.1. General Features of Erotylidae mt Genomes
3.2. Protein-Coding Genes
3.3. Transfer RNAs, Ribosomal RNAs, and Control Region
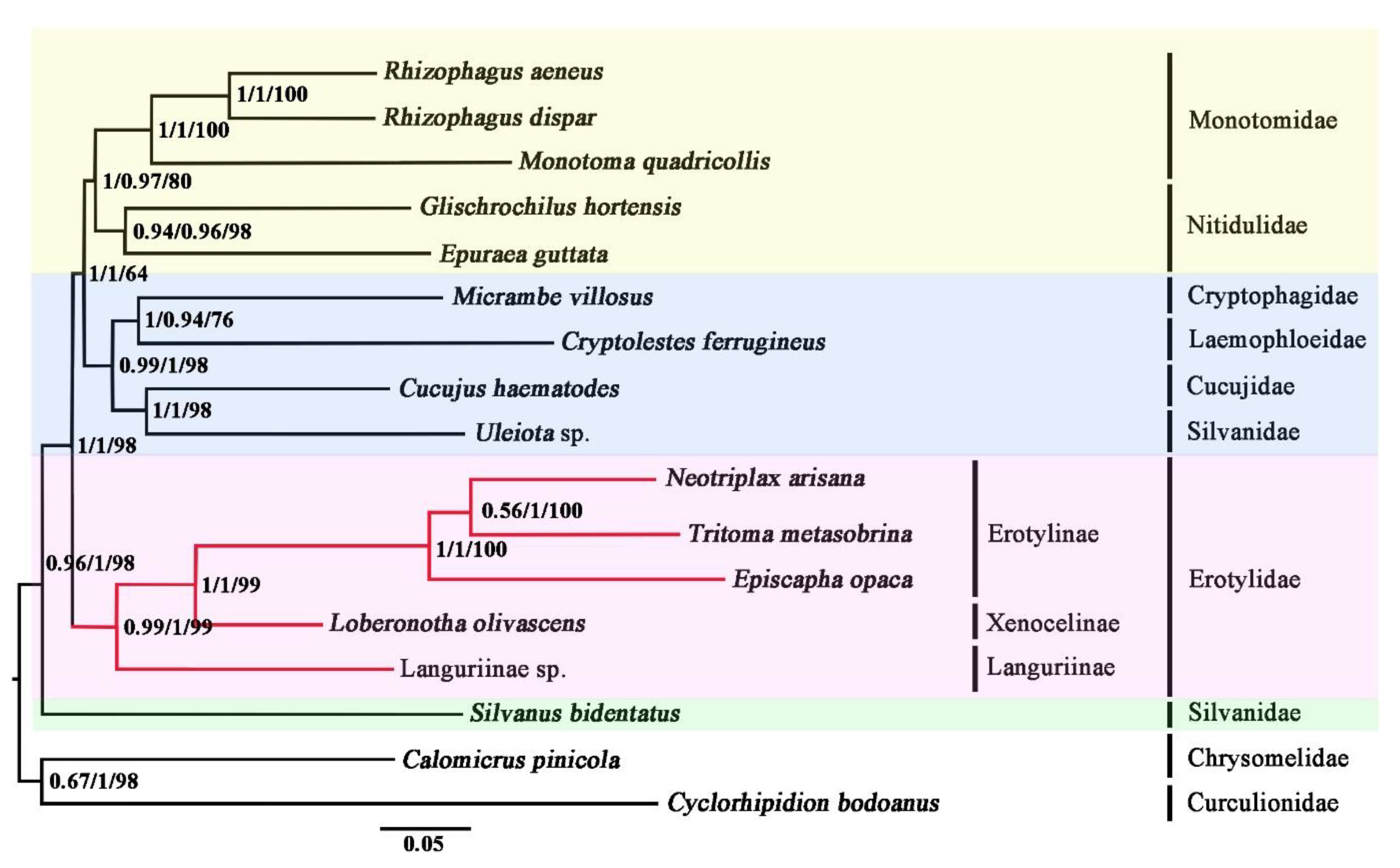
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leschen, R.A.B.; Skelley, P.E.; McHugh, J.V. Erotylidae Leach 1785. In Band/Volume IV arthropoda: Insectateilband/part 38. Coleoptera, beetles, Volume 2. Morphology and systematics (Polyphagapartim); Leschen, R.A.B., Beutel, R.G., Lawrence, J.F., Eds.; Handbuch der Zoologie/Handbook of Zoology; W. DeGruyter: Berlin, Germany, 2010; pp. 311–319. [Google Scholar]
- Leschen, R.A.B.; Buckley, T.R. Multistate Characters and Diet Shifts: Evolution of Erotylidae (Coleoptera). Syst. Biol. 2007, 56, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Latreille, P.A. Histoire Naturelle, Générale et particulière des Crustacéset des Insectes; De L’Imprimere de F. Dufart: Paris, France, 1802; p. 467. [Google Scholar]
- Leschen, R.A.B.; Lawrence, J.F.; Ślipiński, S.A. Classification of basal Cucujoidea (Coleoptera:Polyphaga): Cladistic analysis, keys and review of new families. Invertebr. Syst. 2005, 19, 17–73. [Google Scholar] [CrossRef]
- Lawrence, J.F.; Newton, A.F. Evolution and Classification of Beetles. Annu. Rev. Ecol. Syst. 1982, 13, 261–290. [Google Scholar] [CrossRef] [Green Version]
- Leschen, R.A.B.; Beutel, R.G.; Lawrence, J.F.; Ślipiński, A. Coleoptera, Beetles, Volume 2, Morphology and Systematics (Elateroidea, Bostrichiformia, Cucujiformia partim); Handbook of Zoology; De Gruyter: Berlin, Germany; New York, NY, USA, 2011. [Google Scholar] [CrossRef]
- McElrath, T.; Robertson, J.A.; Thomas, M.C.; Osborne, J.T.; Miller, K.B.; McHugh, J.; Whiting, M.F. A molecular phylogenetic study of Cucujidae s.l. (Coleoptera: Cucujoidea). Syst. Èntomol. 2015, 40, 705–718. [Google Scholar] [CrossRef]
- Hunt, T.; Bergsten, J.; Gómez-Zurita, J.; Ribera, I.; Barraclough, T.G.; Bocakova, M.; Bocak, L.; Vogler, A.P.; Levkanicova, Z.; Papadopoulou, A.; et al. A Comprehensive Phylogeny of Beetles Reveals the Evolutionary Origins of a Superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef]
- Bocak, L.; Barton, C.; Crampton-Platt, A.; Chesters, D.; Ahrens, D.; Vogler, A.P. Building the Coleoptera tree-of-life for >8000 species: Composition of public DNA data and fit with Linnaean classification. Syst. Èntomol. 2013, 39, 97–110. [Google Scholar] [CrossRef]
- Lawrence, J.F.; Ślipiński, A.; Seago, A.E.; Thayer, M.K.; Newton, A.F.; Marvaldi, A.E. Phylogeny of the Coleoptera Based on Morphological Characters of Adults and Larvae. Ann. Zool. 2011, 61, 1–217. [Google Scholar] [CrossRef]
- Crotch, G.R. A revision of the Coleopterous Family Erotylidae. Cistula Entomol. 1876, 1, 359–572. [Google Scholar]
- Leschen, R.A.B. Erotylidae (Insecta: Coleoptera: Cucujoidea): Phylogeny and review. Fauna N. Z. 2003, 47, 108. [Google Scholar]
- Wegrzynowicz, P. Morphology, phylogeny and classification of the family Erotylidae based on adult characters (Coleoptera: Cucujoidea). Genus 2002, 13, 314–319. [Google Scholar] [CrossRef]
- Robertson, J.A.; McHugh, J.V.; Whiting, M.F. A molecular phylogenetic analysis of the pleasing fungus beetles (Coleoptera: Erotylidae): Evolution of colour patterns, gregariousness and mycophagy. Syst. Èntomol. 2004, 29, 173–187. [Google Scholar] [CrossRef]
- Arrow, G.J. Coleoptera. Clavicornia. Erotylidae, Languriidae, and Endomychidae. In The Fauna of British India, including Ceylon and Burma; Taylor and Francis: London, UK, 1925; p. 416. [Google Scholar]
- Crowson, R.A. The Natural Classification of the Families of Coleoptera; Nathaniel Lloyd: London, UK, 1955; p. 187. [Google Scholar]
- Sen, G.T.; Crowson, R.A. A review of classification of the family Languriidae (Coleoptera: Clavicornia) and the place of Languriidae in the natural system of Clavicornia. Mem. Zool. Surv. India 1971, 15, 1–42. [Google Scholar]
- Leschen, R.A.B. Phylogeny and revision of genera of Cryptophagidae (Coleoptera: Cucujoidea). Kans. Sci. Bull. 1996, 55, 549–643. [Google Scholar]
- Drilling, K.; Dettner, K.; Klass, K.-D. Morphology of the pronotal compound glands in Tritoma bipustulata (Coleoptera: Erotylidae). Org. Divers. Evol. 2010, 10, 205–214. [Google Scholar] [CrossRef]
- Wolstenholme, D.R.; Clary, D.O. Sequenece evolution of drosophila mitochondrial DNA. Genetics 1985, 109, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Sankoff, D.; Leduc, G.; Antoine, N.; Paquin, B.; Lang, B.F.; Cedergren, R. Gene order comparisons for phylogenetic inference: Evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. USA 1992, 89, 6575–6579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Mauro, D.S.; Gower, D.J.; Zardoya, R.; Wilkinson, M. A Hotspot of Gene Order Rearrangement by Tandem Duplication and Random Loss in the Vertebrate Mitochondrial Genome. Mol. Biol. Evol. 2005, 23, 227–234. [Google Scholar] [CrossRef]
- Cameron, S.L.; Lo, N.; Bourguignon, T.; Svenson, G.J.; Evans, T. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): Robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol. Phylogenetics Evol. 2012, 65, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Chûjô, M. Descriptions of some new Erotylidae (Coleoptera) from the Japanese Empire. Jpn. J. Entomol. 1941, 15, 10–21. [Google Scholar]
- Miwa, Y. On the Erotylidae of Japan, Formosa, Corea and Saghalien. Trans. Nat. Hist. Soc. Taihoku 1929, 19, 124–127. [Google Scholar]
- Heller, K.M. Beitrag zur Kenntnis der Erotyliden der Indo-Australischen Region mit besonderer Berücksichtigung der Philippinischen Arten. Arch. Nat. 1918, 8, 79–80. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013, 41, W471–W474. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, N.; Kocher, T. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.A.; Ślipiński, A.; Moulton, M.; Shockley, F.; Giorgi, A.; Lord, N.; McKenna, D.D.; Tomaszewska, W.; Forrester, J.; Miller, K.B.; et al. Phylogeny and classification of Cucujoidea and the recognition of a new superfamily Coccinelloidea (Coleoptera: Cucujiformia). Syst. Èntomol. 2015, 40, 745–778. [Google Scholar] [CrossRef]
- Johnson, P.J.; Löbl, I.; Smetana, A. Catalogue of Palaearctic Coleoptera. Volume 3: Scarabaeoidea–Scirtoidea–Dascilloidea–Buprestoidea–Byrrhoidea; and Volume 4: Elateroidea–Derodontoidea–Bostrichoidea–Lymexyloidea–Cleroidea–Cucujoidea. Ann. Èntomol. Soc. Am. 2009, 102, 735–736. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Philippe, H. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Tihelka, E.; Pisani, D.; Donoghue, P.C. Data curation and modeling of compositional heterogeneity in insect phylogenomics: A case study of the phylogeny of Dytiscoidea (Coleoptera: Adephaga). Mol. Phylogenetics Evol. 2020, 147, 106782. [Google Scholar] [CrossRef]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, L.; Ollikainen, A.; Dodsworth, S.; Foster, P.; Bocak, L.; Vogler, A.P. Family-Level Sampling of Mitochondrial Genomes in Coleoptera: Compositional Heterogeneity and Phylogenetics. Genome Biol. Evol. 2016, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. Institute of Evolutionary Biology; University of Edinburgh: Edinburgh, UK, 2009; Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 25 May 2021).
- Nie, R.E.; Yang, X.K. Research progress in mitochondrial genomes of Coleoptera. Acta Entomol. Sin. 2014, 57, 860–868. [Google Scholar]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates ofsubstitution. Mol. Phylogenetics Evol. 2006, 38, 100–116. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Mori, S.; Matsunami, M. Signature of positive selection in mitochondrial DNA in Cetartiodactyla. Genes Genet. Syst. 2018, 93, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Ge, X.; Xie, G.; Liu, H.; Yang, Y. First Complete Mitochondrial Genome of Melyridae (Coleoptera, Cleroidea): Genome Description and Phylogenetic Implications. Insects 2021, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Che, L.-H.; Li, Y.; Liang, D.; Pang, H.; Ślipiński, A.; Zhang, P. Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Accession No. | Size (bp) |
|---|---|---|---|
| Ingroups | |||
| Cryptophagidae | Micrambe villosus | KX087317.1 | 17,907 bp |
| Cucujidae | Cucujus haematodes | KX087268.1 | 16,120 bp |
| Laemophloeidae | Cryptolestes ferrugineus | KT182067.1 | 15,511 bp |
| Monotomidae | Monotoma quadricollis | NC036266.1 | 16,064 bp |
| Monotomidae | Rhizophagus aeneus | KX087340.1 | 16,454 bp |
| Monotomidae | Rhizophagus dispar | KX035133.1 | 13,423 bp |
| Nitidulidae | Epuraea guttata | KX087289.1 | 16,021 bp |
| Nitidulidae | Glischrochilus hortensis | JX412778.1 | 10,677 bp |
| Silvanidae | Silvanus bidentatus | KX035145.1 | 17,220 bp |
| Silvanidae | Uleiota sp. | KX035149.1 | 14,967 bp |
| Erotylidae | Languriinae sp. | MG193464.1 | 16,082 bp |
| Erotylidae | Loberonotha olivascens | JX412784.1 | 13,039 bp |
| Erotylidae | Tritoma metasobrina | MZ014622 | 16,502 bp |
| Erotylidae | Episcapha opaca | MZ014623 | 15,581 bp |
| Erotylidae | Neotriplax arisana | MZ014624 | 16,478 bp |
| Outgroups | |||
| Curculionidae | Cyclorhipidion bodoanus | NC036295.1 | 15,899 bp |
| Chrysomelidae | Calomicrus pinicola | KX087334.1 | 15,908 bp |
| Gene | Strand | Location | Length (bp) | Anticodon | Codon | Intergenic Sequence (bp) | ||
|---|---|---|---|---|---|---|---|---|
| Start | End | Start | Stop | |||||
| tRNAIle | J | 1 | 74/63/67/63 | 74/63/67/63 | CAT | 145/61/−3/2 | ||
| TrnaGln | N | 220/125/65/66 | 288/193/133/134 | 69 | TTG | 3/0/3/3 | ||
| tRNAMet | J | 292/194/137/138 | 360/262/205/206 | 69 | GAT | 33/18/18/21 | ||
| ND2 | J | 394/281/224/228 | 1369/1271/1217/1221 | 976/991/994/994 | ATA/ATA/ATC/ATA | T-tRNA | 0 | |
| tRNATrp | J | 1370/1272/1218/1222 | 1437/1334/1284/1287 | 68/63/67/66 | TCA | 0/−8/−1/−1 | ||
| tRNACys | N | 1438/1327/1284/1287 | 1501/1389/1348/1350 | 64/63/65/64 | GCA | 21/0/1/2 | ||
| tRNATyr | N | 1523/1390/1350/1353 | 1588/1452/1414/1418 | 66/63/65/66 | GTA | −8/0/1/10 | ||
| COXI | J | 1581/1453/1416/1429 | 3120/2987/2946/2950 | 1540/1535/1531/1522 | ATT/TTG/ATT/TTG | T-tRNA | 0 | |
| tRNALeu(UUR) | J | 3121/2988/2947/2951 | 3185/3051/3011/3015 | 65/64/65/65 | TAA | 18/0/18/0 | ||
| COXII | J | 3204/3052/3030/3016 | 3876/3738/3695/3700 | 673/687/666/685 | ATA/ATT/ATT/ATT | T-tRNA/TAA/TAA/T-tRNA | 0/1/2/0 | |
| tRNALys | J | 3877/3740/3698/3701 | 3947/3809/3767/3770 | 71/70/70/70 | CTT | −1/−1/0/0 | ||
| tRNAAsp | J | 3947/3809/3768/3771 | 4012/3873/3833/3836 | 66/65/66/66 | GTC | 25/18/32/0 | ||
| ATP8 | J | 4038/3892/3866/3837 | 4193/4047/4021/3992 | 156 | ATT/ATT/ATT/ATC | TAA/TAA/TAA/TAG | −4/−7/−4/−7 | |
| ATP6 | J | 4190/4041/4018/3986 | 4855/4712/4683/4657 | 666/672/666/672 | ATA/ATG/ATA/ATG | TAA | −1 | |
| COXIII | J | 4855/4712/4683/4657 | 5642/5498/5469/5445 | 788/787/787/789 | ATG | TA-tRNA/T-tRNA/T-tRNA/TAA | 0/0/0/6 | |
| tRNAGly | J | 5643/5499/5470/5452 | 5708/5561/5534/5517 | 66/63/65/66 | TCC | 9/30/12/0 | ||
| ND3 | J | 5718/5592/5547/5518 | 6062/5913/5886/5871 | 345/322/340/354 | ATA/ATA/ATA/ATT | TAA/T-tRNA/T-tRNA/TAA | 5/0/0/2 | |
| tRNAAla | J | 6068/5914/5887/5874 | 6132/5980/5955/5939 | 65/67/69/66 | TGC | −1 | ||
| tRNAArg | J | 6132/5980/5955/5939 | 6196/6042/6018/6002 | 65/63/64/64 | TCG | −1 | ||
| tRNAAsn | J | 6196/6042/6018/6002 | 6259/6105/6081/6067 | 64/64/64/66 | GTT | 0 | ||
| tRNASer(AGN) | J | 6260/6106/6082/6068 | 6327/6171/6148/6134 | 68/66/67/67 | GCT | 1/0/0/0 | ||
| tRNAGlu | J | 6329/6172/6149/6135 | 6392/6233/6213/6197 | 64/62/65/63 | TTC | −2/−2/−2/15 | ||
| tRNAPhe | N | 6391/6232/6212/6213 | 6455/6293/6277/6277 | 65/62/66/65 | GAA | 0 | ||
| ND5 | N | 6456/6294/6278/6278 | 8124/7968/7901/7991 | 1669/1675/1624/1714 | ATT/ATA/ATA/GTG | T-tRNA | 45/39/90/0 | |
| tRNAHis | N | 8170/8008/7992/7992 | 8233/8068/8055/8055 | 64/61/64/64 | GTG | 0 | ||
| ND4 | N | 8234/8069/8056/8056 | 9568/9401/9388/9388 | 1335/1333/1333/1333 | ATG | TAA/T-tRNA/T-tRNA/T-tRNA | −7 | |
| ND4L | N | 9562/9395/9382/9382 | 9843/9676/9663/9669 | 282/282/282/288 | ATT/ATA/ATT/ATG | TAA | 14/8/8/2 | |
| tRNAThr | J | 9858/9685/9672/6972 | 9921/9746/9738/9734 | 64/62/67/63 | TGT | 0 | ||
| tRNAPro | N | 9922/9747/9739/9735 | 9986/9811/9803/9798 | 65/65/65/64 | TGG | 4/1/1/5 | ||
| ND6 | J | 9991/9813/9805/9804 | 10,491/10,313/10,311/10,307 | 501/501/507/504 | ATA | TAA | 6/11/5/−1 | |
| CytB | J | 10,498/10,325/10,317/10,307 | 11,629/11,453/11,452/11,446 | 1132/1129/1136/1140 | ATA/ATT/ATA/ATG | T-tRNA/T-tRNA/TA-tRNA/TAA | 0 | |
| tRNASer(UCN) | J | 11,630/11,454/11,453/11,447 | 11,697/11,520/11,519/11,513 | 68/67/67/67 | TGA | 16/16/25/24 | ||
| ND1 | N | 11,714/11,537/11,545/11,538 | 12,664/12,487/12,495/12,488 | 951 | TTG/TTG/TTG/ATT | TAG | 1 | |
| tRNALeu(CUN) | N | 12,666/12,489/12,497/12,490 | 12,732/12,550/12,561/12,554 | 67/62/65/65 | TAG | 0/29/0/0 | ||
| rrnL | N | 12,733/12,580/12,562/12,555 | 14,009/13,631/13,846/13,933 | 1365/1052/1285/1379 | 0/197/0/0 | |||
| tRNAVal | N | 14,010/13,829/13,847/13,934 | 14,079/13,893/13,916/14,003 | 70/65/70/70 | TAC | 0/0/0/0 | ||
| rrnS | N | 14,080/13,894/13,917/14,004 | 14,850/14,652/14,693/14,777 | 772/759/777/774 | 0 | |||
| A + T rich-region | 14,851/14,653/14,694/14,778 | 16,502/15,581/16,478/16,082 | 1652/929/1785/1305 | 0 | ||||
| Species | CG | PCGs | 16S rRNA | 12S rRNA | CR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A + T (%) | AT Skew | GC Skew | A + T (%) | AT Skew | GC Skew | A + T (%) | AT Skew | GC Skew | A + T (%) | AT Skew | GC Skew | A + T (%) | AT Skew | GC Skew | |
| T. metasobrina | 74.80 | 0.04 | −0.29 | 72.90 | 0.04 | −0.25 | 81.70 | 0.05 | −0.31 | 77.90 | 0.05 | −0.34 | 75.50 | 0.04 | −0.66 |
| E. opaca | 78.70 | 0.03 | −0.20 | 77.79 | −0.12 | −0.01 | 80.70 | −0.04 | 0.31 | 81.03 | 0.00 | 0.32 | 81.59 | 0.17 | −0.27 |
| N. arisana | 75.39 | 0.04 | −0.23 | 73.66 | −0.15 | −0.02 | 81.48 | −0.01 | 0.34 | 79.43 | 0.00 | 0.31 | 77.54 | 0.12 | −0.50 |
| * Languriinae sp. | 76.46 | −0.01 | −0.20 | 74.96 | −0.15 | −0.02 | 81.22 | 0.07 | −0.38 | 78.55 | −0.01 | −0.31 | 79.69 | −0.02 | −0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, Y.; Zhang, R.; Shi, C.; Lu, W.; Li, J.; Bai, M. Three Complete Mitochondrial Genomes of Erotylidae (Coleoptera: Cucujoidea) with Higher Phylogenetic Analysis. Insects 2021, 12, 524. https://doi.org/10.3390/insects12060524
Liu J, Wang Y, Zhang R, Shi C, Lu W, Li J, Bai M. Three Complete Mitochondrial Genomes of Erotylidae (Coleoptera: Cucujoidea) with Higher Phylogenetic Analysis. Insects. 2021; 12(6):524. https://doi.org/10.3390/insects12060524
Chicago/Turabian StyleLiu, Jing, Yuyu Wang, Ruyue Zhang, Chengmin Shi, Weicheng Lu, Jing Li, and Ming Bai. 2021. "Three Complete Mitochondrial Genomes of Erotylidae (Coleoptera: Cucujoidea) with Higher Phylogenetic Analysis" Insects 12, no. 6: 524. https://doi.org/10.3390/insects12060524
APA StyleLiu, J., Wang, Y., Zhang, R., Shi, C., Lu, W., Li, J., & Bai, M. (2021). Three Complete Mitochondrial Genomes of Erotylidae (Coleoptera: Cucujoidea) with Higher Phylogenetic Analysis. Insects, 12(6), 524. https://doi.org/10.3390/insects12060524