
Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
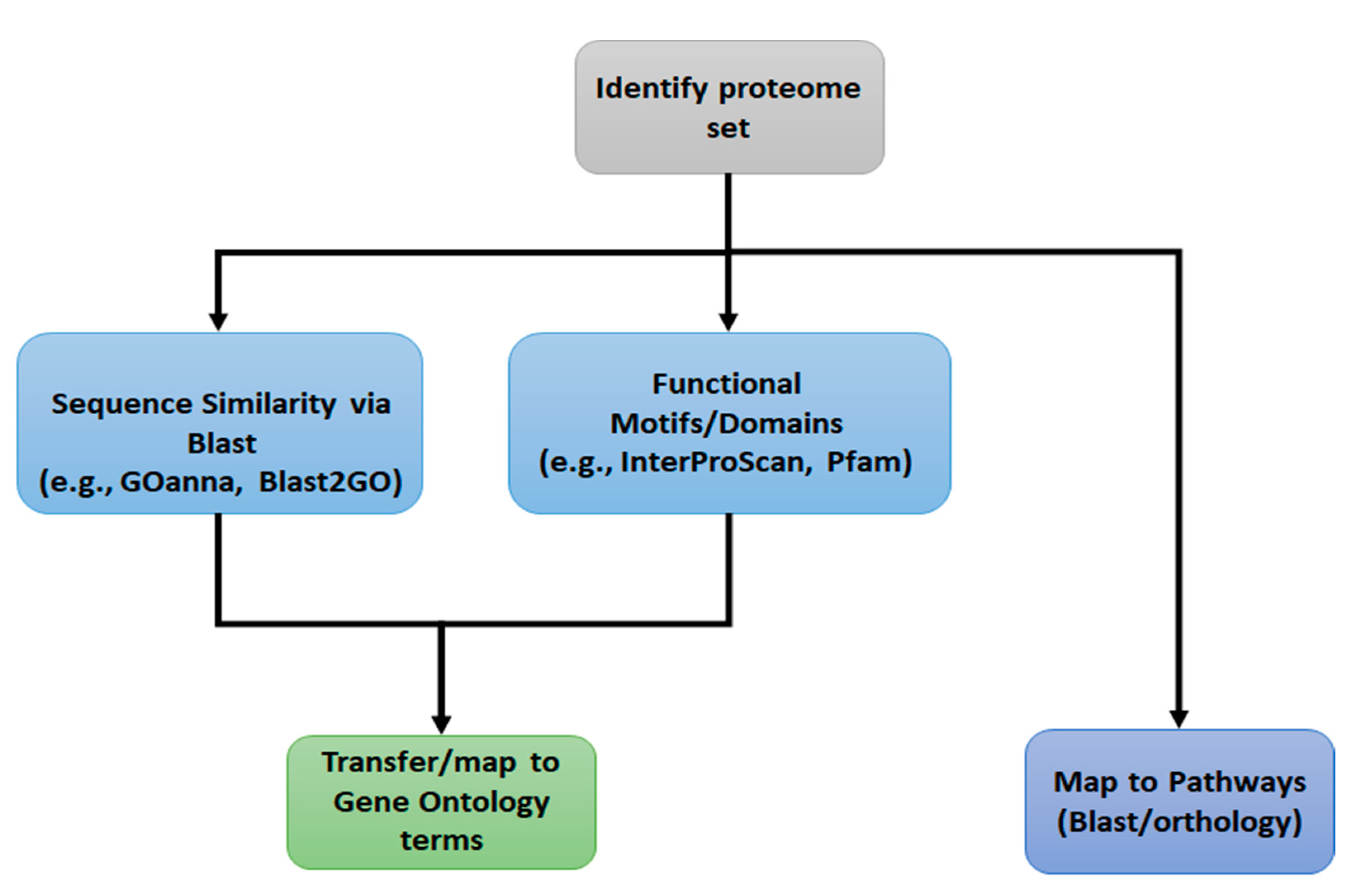
Motivation
2. Materials and Methods
2.1. Sequence Similarity via BLAST: GOanna
2.2. Functional Motif Analysis: InterProScan
2.3. Combining and QC of GO Annotations
2.4. Map to Pathways: KOBAS
- Annotate: This step assigns appropriate KEGG Ortholog (KO) terms for queried sequences based on a similarity search. It also assigns proteins to pathways from KEGG, Reactome, and BioCyc.
- Identify: This performs an enrichment analysis compared to a background of the species’ gene set among the annotation results based on the frequency or statistical significance of pathways.
2.5. Research Design and Method: Comparing Functional Annotation across Multiple Species
3. Results
3.1. Installation and Runtime Considerations
3.2. Parameter Optimization
3.3. Results and Discussion
3.3.1. Genome Assembly
3.3.2. Gene Ontology Annotation
3.3.3. Pathway Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buza, T.J.; Kumar, R.; Gresham, C.R.; Burgess, S.C.; McCarthy, F.M. Facilitating Functional Annotation of Chicken Microarray Data. BMC Bioinform. 2009, 10 (Suppl. 11), S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P.; et al. AgBase: A Functional Genomics Resource for Agriculture. BMC Genom. 2006, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-Based Propagation of Functional Annotations within the Gene Ontology Consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, M.J. Phylogenetic Signal in the Eukaryotic Tree of Life. Science 2008, 321, 121–123. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.M.; Gresham, C.R.; Buza, T.J.; Chouvarine, P.; Pillai, L.R.; Kumar, R.; Ozkan, S.; Wang, H.; Manda, P.; Arick, T.; et al. AgBase: Supporting Functional Modeling in Agricultural Organisms. Nucleic Acids Res. 2011, 39, D497–D506. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant. Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, G.; Götz, S.; Conesa, A.; Segrelles, D.; Blanquer, I.; García, J.M.; Hernandez, V.; Robles, M.; Talon, M. Blast2GO Goes Grid: Developing a Grid-Enabled Prototype for Functional Genomics Analysis. Stud. Health Technol. Inform. 2006, 120, 194–204. [Google Scholar] [PubMed]
- Balakrishnan, R.; Harris, M.A.; Huntley, R.; Van Auken, K.; Cherry, J.M. A Guide to Best Practices for Gene Ontology (GO) Manual Annotation. Database 2013, 2013, bat054. [Google Scholar] [CrossRef] [PubMed]
- Ag100Pest Species. Available online: http://i5k.github.io/ag100pest (accessed on 9 June 2021).
- Childers, A.K.; Geib, S.M.; Sim, S.B.; Poelchau, M.F. The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects 2021, 12, 626. [Google Scholar] [CrossRef] [PubMed]
- Lewin, H.A.; Robinson, G.E.; Kress, W.J.; Baker, W.J.; Coddington, J.; Crandall, K.A.; Durbin, R.; Edwards, S.V.; Forest, F.; Gilbert, M.T.P.; et al. Earth BioGenome Project: Sequencing Life for the Future of Life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Buza, T.J.; McCarthy, F.M.; Wang, N.; Bridges, S.M.; Burgess, S.C. Gene Ontology Annotation Quality Analysis in Model Eukaryotes. Nucleic Acids Res. 2008, 36, e12. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, F.M.; Bridges, S.M.; Wang, N.; Magee, G.B.; Williams, W.P.; Luthe, D.S.; Burgess, S.C. AgBase: A Unified Resource for Functional Analysis in Agriculture. Nucleic Acids Res. 2007, 35, D599–D603. [Google Scholar] [CrossRef] [Green Version]
- Poelchau, M.; Childers, C.; Moore, G.; Tsavatapalli, V.; Evans, J.; Lee, C.-Y.; Lin, H.; Lin, J.-W.; Hackett, K. The i5k Workspace@NAL--Enabling Genomic Data Access, Visualization and Curation of Arthropod Genomes. Nucleic Acids Res. 2015, 43, D714–D719. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.A.; Vaughn, M.; McKay, S.; Lyons, E.; Stapleton, A.E.; Gessler, D.; Matasci, N.; Wang, L.; Hanlon, M.; Lenards, A.; et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front. Plant. Sci. 2011, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devisetty, U.K.; Kennedy, K.; Sarando, P.; Merchant, N.; Lyons, E. Bringing Your Tools to CyVerse Discovery Environment Using Docker. F1000Res. 2016, 5, 1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Functional Annotation Workflow—AgBase 1.0 Documentation. Available online: https://agbase-docs.readthedocs.io/en/latest/agbase/workflow.html (accessed on 9 June 2021).
- TransDecoder TransDecoder/TransDecoder. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 22 May 2021).
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A Web Server for Annotation and Identification of Enriched Pathways and Diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-I: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Index of /Data/Arthropoda/Apimel-(Apis_Mellifera)/Amel_4.5/2.Official or Primary Gene Set/Apimel_OGSv3.3/. Available online: https://i5k.nal.usda.gov/data/Arthropoda/apimel-%28Apis_mellifera%29/Amel_4.5/2.Official%20or%20Primary%20Gene%20Set/apimel_OGSv3.3/ (accessed on 10 June 2021).
- Index of /Releases/FB2020_05/Dmel_r6.36/Fasta. Available online: http://ftp.flybase.net/releases/FB2020_05/dmel_r6.36/fasta/ (accessed on 10 June 2021).
- Index of /Data/Arthropoda/Tricas-(Tribolium_Castaneum)/Current Genome Assembly/2.Official or Primary Gene Set/TCAS_OGS_v3/. Available online: https://i5k.nal.usda.gov/data/Arthropoda/tricas-%28Tribolium_castaneum%29/Current%20Genome%20Assembly/2.Official%20or%20Primary%20Gene%20Set/TCAS_OGS_v3/ (accessed on 10 June 2021).
- Richards, S.; Hughes, D.; Ayoub, N. Latrodectus hesperus Genome Annotations v0.5.3 2019. Available online: https://i5k.nal.usda.gov/data/Arthropoda/lathes-(Latrodectus_hesperus)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/consensus_gene_set/LHES.faa (accessed on 18 August 2021).
- Richards, S.; Hughes, D.; Niehuis, O. Limnephilus lunatus Genome Annotations v0.5.3 2019. Available online: https://i5k.nal.usda.gov/data/Arthropoda/limlun-(Limnephilus_lunatus)/Current%20Genome%20Assembly/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/consensus_gene_set/LLUN.faa (accessed on 18 August 2021).
- Panfilio, K.; Richards, S.; Viala, S.; van der Zee, M.; Traverso, L.; Tidswell, O.; Suzuki, Y.; Shukla, J.; Sghaier, E.; Seibert, J.; et al. Oncopeltus fasciatus Official Gene Set v1.2 2020. Available online: https://i5k.nal.usda.gov/data/Arthropoda/oncfas-(Oncopeltus_fasciatus)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/OGS_v1.2/oncfas_OGSv1.2_02192016/oncfas_OGSv1.2_original_peptide.fa (accessed on 18 August 2021).
- Hughes, D.S.T.; Hunter, W.B.; Richards, S. Homalodisca vitripennis Genome Annotations v0.5.3 2015. Available online: https://i5k.nal.usda.gov/data/Arthropoda/homvit-(Homalodisca_vitripennis)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/consensus_gene_set/HVIT.faa (accessed on 18 August 2021).
- Index of /Data/Arthropoda/Euraff-(Eurytemora_Affinis Complex (Atlantic Clade))/BCM-After-Atlas/2.Official or Primary Gene Set/BCM_Version_0.5.3/. Available online: https://i5k.nal.usda.gov/data/Arthropoda/euraff-%28Eurytemora_affinis%20complex%20%28Atlantic%20clade%29%29/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/ (accessed on 15 July 2021).
- Richards, S.; Hughes, D.; Kuhn, K.; Duan, J. Agrilus planipennis Genome Annotations v0.5.3 2019. Available online: https://i5k.nal.usda.gov/data/Arthropoda/agrpla-(Agrilus_planipennis)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/consensus_gene_set/APLA.faa (accessed on 18 August 2021).
- Richards, S.; Hughes, D.; Strand, M. Copidosoma floridanum Genome Annotations v0.5.3 2019. Available online: https://i5k.nal.usda.gov/data/Arthropoda/copflo-(Copidosoma_floridanum)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/BCM_version_0.5.3/consensus_gene_set/CFLO.faa (accessed on 18 August 2021).
- Oeyen, J.P.; Hatakeyama, M.; Hughes, D.S.T.; Richards, S.; Misof, B.; Niehuis, O. Athalia rosae Genome Annotations v0.5.3 2018. Available online: https://i5k.nal.usda.gov/data/Arthropoda/athros-(Athalia_rosae)/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/athros_OGS_v1.0/ATHROS_OGSv1-0_pep.fa (accessed on 18 August 2021).
- Index of /Data/Arthropoda/Cercap-(Ceratitis_Capitata)/GCF_000347755.1/2.Official or Primary Gene Set/OGSv1/. Available online: https://i5k.nal.usda.gov/data/Arthropoda/cercap-%28Ceratitis_capitata%29/GCF_000347755.1/2.Official%20or%20Primary%20Gene%20Set/OGSv1/ (accessed on 15 July 2021).
- Index of /Data/Arthropoda/Cimlec-(Cimex_Lectularius)/BCM-After-Atlas/2.Official or Primary Gene Set/OGS_v1_2/. Available online: https://i5k.nal.usda.gov/data/Arthropoda/cimlec-%28Cimex_lectularius%29/BCM-After-Atlas/2.Official%20or%20Primary%20Gene%20Set/OGS_v1_2/ (accessed on 15 July 2021).
- Website. Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/Varroa_destructor/protein/protein.fa.gz (accessed on 10 June 2021).
- Hosmani, P.S.; Flores-Gonzalez, M.; Shippy, T.; Vosburg, C.; Massimino, C.; Tank, W.; Reynolds, M.; Tamayo, B.; Miller, S.; Norus, J.; et al. Chromosomal Length Reference Assembly for Diaphorina Citri Using Single-Molecule Sequencing and Hi-C Proximity Ligation with Manually Curated Genes in Developmental, Structural and Immune Pathways. bioRxiv 2019, 869685. [Google Scholar] [CrossRef] [Green Version]
- InterProScan on the Command Line—AgBase 1.0 Documentation. Available online: https://agbase-docs.readthedocs.io/en/latest/interproscan/using_iprs_cmd.html (accessed on 15 July 2021).
- European Bioinformatics Institute. Available online: https://www.ebi.ac.uk/GOA/fly_release (accessed on 27 May 2021).
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc Collection of Microbial Genomes and Metabolic Pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Dunn, N.A.; Unni, D.R.; Diesh, C.; Munoz-Torres, M.; Harris, N.L.; Yao, E.; Rasche, H.; Holmes, I.H.; Elsik, C.G.; Lewis, S.E. Apollo: Democratizing Genome Annotation. PLoS Comput. Biol. 2019, 15, e1006790. [Google Scholar] [CrossRef] [Green Version]
- Giraldo-Calderón, G.I.; Emrich, S.J.; MacCallum, R.M.; Maslen, G.; Dialynas, E.; Topalis, P.; Ho, N.; Gesing, S.; VectorBase Consortium; Madey, G.; et al. VectorBase: An Updated Bioinformatics Resource for Invertebrate Vectors and Other Organisms Related with Human Diseases. Nucleic Acids Res. 2015, 43, D707–D713. [Google Scholar] [CrossRef]
- Elsik, C.G.; Tayal, A.; Diesh, C.M.; Unni, D.R.; Emery, M.L.; Nguyen, H.N.; Hagen, D.E. Hymenoptera Genome Database: Integrating Genome Annotations in HymenopteraMine. Nucleic Acids Res. 2016, 44, D793–D800. [Google Scholar] [CrossRef] [Green Version]
- Flores-Gonzalez, M.; Hosmani, P.S.; Fernandez-Pozo, N.; Mann, M.; Humann, J.L.; Main, D.; Heck, M.; Brown, S.J.; Mueller, L.A.; Saha, S. Citrusgreening.org: An Open Access and Integrated Systems Biology Portal for the Huanglongbing (HLB) Disease Complex. bioRxiv 2019, 868364. [Google Scholar] [CrossRef] [Green Version]
- Hosmani, P.S.; Shippy, T.; Miller, S.; Benoit, J.B.; Munoz-Torres, M.; Flores-Gonzalez, M.; Mueller, L.A.; Wiersma-Koch, H.; D’Elia, T.; Brown, S.J.; et al. A Quick Guide for Student-Driven Community Genome Annotation. PLoS Comput. Biol. 2019, 15, e1006682. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Shippy, T.D.; Hosmani, P.S.; Flores-Gonzalez, M.; Mueller, L.A.; Hunter, W.B.; Brown, S.J.; D’elia, T.; Saha, S. Segmentation Pathway Genes in the Asian Citrus Psyllid, Diaphorina citri. bioRxiv 2020. [Google Scholar] [CrossRef]
- Miller, S.; Shippy, T.D.; Tamayo, B.; Hosmani, P.S.; Flores-Gonzalez, M.; Mueller, L.A.; Hunter, W.B.; Brown, S.J.; D’elia, T.; Saha, S. Characterization of Chitin Deacetylase Genes in the Diaphorina citri Genome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vosburg, C.; Reynolds, M.; Noel, R.; Shippy, T.; Hosmani, P.S.; Flores-Gonzalez, M.; Mueller, L.A.; Hunter, W.B.; Brown, S.J.; D’Elia, T.; et al. Utilizing a Chromosomal-Length Genome Assembly to Annotate the Wnt Signaling Pathway in the Asian Citrus Psyllid, Diaphorina citri. Gigabyte 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Massimino, C.; Vosburg, C.; Shippy, T.; Hosmani, P.S.; Flores-Gonzalez, M.; Mueller, L.A.; Hunter, W.B.; Benoit, J.B.; Brown, S.J.; D’Elia, T.; et al. Annotation of Yellow Genes in Diaphorina citri, the Vector for Huanglongbing Disease. Gigabyte 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Miller, S.; Shippy, T.D.; Tamayo, B.; Hosmani, P.S.; Flores-Gonzalez, M.; Mueller, L.A.; Hunter, W.B.; Brown, S.J.; D’Elia, T.; Saha, S. Annotation of Chitin Biosynthesis Genes in Diaphorina citri, the Asian Citrus Psyllid. Gigabyte 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Docker Hub. Available online: https://hub.docker.com/r/agbase/goanna (accessed on 9 June 2021).
- Docker Hub. Available online: https://hub.docker.com/r/agbase/interproscan (accessed on 9 June 2021).
- Docker Hub. Available online: https://hub.docker.com/r/agbase/combine_gafs (accessed on 9 June 2021).
- Docker Hub. Available online: https://hub.docker.com/r/agbase/kobas (accessed on 9 June 2021).
- Saha, S.; Amcooksey. AgBase/GOanna: AgBase GOanna, Zenodo. 2021. [CrossRef]
- Saha, S.; Amcooksey. AgBase/InterProScan: AgBase InterProScan 5.45-80, Zenodo. 2021. [CrossRef]
- Saha, S.; Amcooksey. AgBase/kobas: AgBase KOBAS 3.0.3, Zenodo. 2021. [CrossRef]
- Ebi-Pf-Team Ebi-Pf-Team/Interproscan. Available online: https://github.com/ebi-pf-team/interproscan (accessed on 9 June 2021).
- KEGG Organisms: Complete Genomes. Available online: https://www.kegg.jp/kegg/catalog/org_list.html (accessed on 9 June 2021).


{kind=link}
| Database Name | No. UniProtKB Proteins | No. in GOanna Db |
|---|---|---|
| arthropod | 3,956,843 | 12,081 |
| bacteria | 28,660,834 | 12,748 |
| bird | 777,091 | 1379 |
| fish | 1,505,807 | 12,478 |
| fungi | 7,614,812 | 13,718 |
| human | 161,566 | 21,125 |
| insecta | 2,883,005 | 11,886 |
| invertebrates | 8,409,505 | 20,741 |
| mammals | 1,836,549 | 42,966 |
| nematode | 1,541,602 | 4941 |
| plants | 6,300,920 | 16,058 |
| UniProt-SwissProt | 50,258 | 72,337 |
| UniProt-TrEMBL | 4,720,107 | 57,834 |
| Database | Description |
|---|---|
| TIGRFAM | TIGRFAMs are protein families based on Hidden Markov Models or HMMs. |
| SFLD | SFLDs are protein families based on Hidden Markov Models or HMMs. |
| ProDom | ProDom is a comprehensive set of protein domain families automatically generated from the UniProt Knowledge Database. |
| Hamap | High-quality Automated and Manual Annotation of Microbial Proteomes. |
| SMART | SMART identifies and analyzes domain architectures based on Hidden Markov Models or HMMs. |
| CDD | Prediction of CDD domains in proteins. |
| ProSiteProfiles | PROSITE consists of documentation entries describing protein domains, families, and functional sites, as well as associated patterns and profiles to identify them. |
| ProSitePatterns | PROSITE consists of documentation entries describing protein domains, families, and functional sites, as well as associated patterns and profiles to identify them. |
| SUPERFAMILY | SUPERFAMILY is a database of structural and functional annotation for all proteins and genomes. |
| PRINTS | A fingerprint is a group of conserved motifs used to characterize a protein family. |
| PANTHER | The PANTHER (protein analysis through evolutionary relationships) Classification System is a unique resource that classifies genes by their functions, using published scientific experimental evidence and evolutionary relationships to predict function even in the absence of direct experimental evidence. |
| Gene3D | Structural assignment for whole genes and genomes using the CATH domain structure database. |
| PIRSF | The PIRSF concept is being used as a guiding principle to provide comprehensive and non-overlapping clustering of UniProtKB sequences into a hierarchical order to reflect their evolutionary relationships. |
| Pfam | A large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs). |
| Coils | Prediction of Coiled Coil Regions in proteins. |
| MobiDBLite | Prediction of disordered domains regions in proteins. |
| Species | Genome Assembly Accession | Genome Assembly Name | Contig N50 | Scaffold N50 | Annotation Name | Proteins | Proteins Assigned GO Terms | Source |
|---|---|---|---|---|---|---|---|---|
| Apis mellifera (honey bee) | GCA_000002195.1 | Amel_4.5 | 5,832,476 | 13,619,445 | OGSv3.3 | 15,314 | 39.91% | [29] |
| Drosophila melanogaster (fruit fly) | GCA_000001215.4 | DMEL_r6.36 | 21,485,538 | 25,286,936 | FB2020_05 | 30,724 | 59.42% | [30] |
| Tribolium castaneum (red flour beetle) | GCA_000002335.3 | TCAS_5.2 | 73,049 | 4,456,720 | TCAS_OGS_v3 | 18,534 | 44.98% | [31] |
| Latrodectus hesperus (Western black widow spider) | GCA_000697925.1 | Lhes_1.0 | 2223 | 13,889 | LHES-BCM_version_0.5.3 | 17,364 | 31.17% | [32] |
| Limnephilus lunatus (caddisfly) | GCA_000648945.1 | Llun_1.0 | 2103 | 54,650 | LLUN-BCM_version_0.5.3 | 13,292 | 55.76% | [33] |
| Oncopeltus fasciatus (large milkweed bug) | GCA_000696205.1 | Ofas_1.0 | 4047 | 339,960 | oncfas_OGSv1.2 | 19,793 | 34.31% | [34] |
| Homalodisca vitripennis (glassy-winged sharpshooter) | GCA_000696855.1 | Hvit_1.0 | 4857 | 512,049 | HVIT-BCM_version_0.5.3 | 33,019 | 38.00% | [35] |
| Eurytemora affinis (calanoid copepod) | GCA_000591075.1 | Eaff_1.0 | 5738 | 862,645 | EAFF-BCM_version_0.5.3 | 29,783 | 30.02% | [36] |
| Agrilus planipennis (emerald ash borer) | GCA_000699045.1 | Apla_1.0 | 6314 | 910,924 | APLA-BCM_version_0.5.3 | 15,497 | 51.07% | [37] |
| Copidosoma floridanum (parasitoid wasp) | GCA_000648655.1 | Cflo_1.0 | 14,521 | 1,037,125 | CFLO-BCM_version_0.5.3 | 19,869 | 34.14% | [38] |
| Athalia rosae (turnip sawfly) | GCA_000344095.1 | Aros_1.0 | 51,418 | 1,366,867 | AROS-BCM_version_0.5.3 | 22,213 | 57.05% | [39] |
| Ceratitis capitata (Mediterranean fruit fly) | GCA_000347755.2 | Ccap_1.1 | 45,879 | 4,118,346 | Ccap-OGSv1 | 12,318 | 55.75% | [40] |
| Cimex lectularius (Cimicidae bed bug) | GCA_000648675.1 | Clec_1.0 | 23,511 | 7,172,596 | Clec-OGSv1.2 | 14,212 | 49.42% | [41] |
| Varroa destructor (parasitic mite) | GCA_002443255.1 | Vdes_3.0 | 201,886 | 58,536,683 | NCBI Varroa destructor Annotation Release 100 | 30,221 | 53.60% | [42] |
| Diaphorina citri (Asian citrus psyllid) | NA | Version 3 | 749,525 | 40,596,296 | OGSv3 | 19,049 | 59.30% | [43] |
| Species | Complete | Complete Single-Copy | Complete Duplicated | Fragmented | Missing |
|---|---|---|---|---|---|
| Drosophila melanogaster (fruit fly) | 99.90 | 53.3 | 46.6 | 0 | 0.1 |
| Athalia rosae (turnip sawfly) | 99.70 | 68.9 | 30.8 | 0 | 0.3 |
| Ceratitis capitata (Mediterranean fruit fly) | 98.40 | 97.5 | 0.9 | 0.4 | 1.2 |
| Tribolium castaneum (red flour beetle) | 98.40 | 93.1 | 5.3 | 1.2 | 0.4 |
| Apis mellifera (honey bee) | 97.40 | 96.9 | 0.5 | 1.5 | 1.1 |
| Varroa destructor (parasitic mite) | 95.90 | 43.1 | 52.8 | 0.7 | 3.4 |
| Cimex lectularius (Cimicidae bed bug) | 95.30 | 93.5 | 1.8 | 2.5 | 2.2 |
| Copidosoma floridanum (parasitoid wasp) | 93.70 | 92.5 | 1.2 | 2.9 | 3.4 |
| Agrilus planipennis (emerald ash borer) | 90.90 | 89.1 | 1.8 | 4.6 | 4.5 |
| Diaphorina citri (Asian citrus psyllid) | 87.10 | 55.9 | 31.2 | 2.8 | 10.1 |
| Oncopeltus fasciatus (large milkweed bug) | 72.90 | 70.8 | 2.1 | 21.4 | 5.7 |
| Eurytemora affinis (calanoid copepod) | 57.50 | 55.9 | 1.6 | 20 | 22.5 |
| Homalodisca vitripennis (glassy-winged sharpshooter) | 55.90 | 54.2 | 1.7 | 32.5 | 11.6 |
| Limnephilus lunatus (caddisfly) | 42.40 | 41.4 | 1 | 28.1 | 29.5 |
| Latrodectus hesperus (Western black widow spider) | 31.40 | 30.6 | 0.8 | 26.9 | 41.7 |
| Species | Proteins | Proteins Assigned GO Terms | GOanna (BLAST) | InterProScan (Motif Analysis) | ||
|---|---|---|---|---|---|---|
| Proteins Assigned GO | Average GAQ | Proteins Assigned GO | Average GAQ | |||
| Apis mellifera (honey bee) | 15,314 | 39.91% | 2.59% | 164.796 | 39.32% | 33.745 |
| Drosophila melanogaster (fruit fly) | 30,724 | 59.42% | 14.85% | 142.024 | 53.12% | 34.847 |
| Tribolium castaneum (red flour beetle) | 18,534 | 44.98% | 2.64% | 142.27 | 44.36% | 33.585 |
| Diaphorina citri (Asian citrus psyllid) | 19,049 | 59.30% | 2.23% | 168.358 | 57.46% | 34.44 |
| Athalia rosae (turnip sawfly) | 22,213 | 57.05% | 2.11% | 144.594 | 56.67% | 35.317 |
| Varroa destructor (parasitic mite) | 30,221 | 53.60% | 0.52% | 167.385 | 53.53% | 33.704 |
| Agrilus planipennis (emerald ash borer) | 15,497 | 51.07% | 2.87% | 179.869 | 41.27% | 31.368 |
| Ceratitis capitata (Mediterranean fruit fly) | 14,212 | 49.42% | 7.94% | 127.988 | 46.42% | 32.504 |
| Cimex lectularius (Cimicidae bed bug) | 14,212 | 49.26% | 3.00% | 177.746 | 48.33% | 35.017 |
| Limnephilus lunatus (caddisfly) | 13,292 | 44.61% | 4.09% | 172.298 | 43.03% | 31.353 |
| Homalodisca vitripennis (glassy-winged sharpshooter) | 33,019 | 38.00% | 1.53% | 174.869 | 30.22% | 30.751 |
| Oncopeltus fasciatus (large milkweed bug) | 19,793 | 34.31% | 2.73% | 189.411 | 33.24% | 29.997 |
| Copidosoma floridanum (parasitoid wasp) | 19,869 | 34.14% | 1.98% | 168.485 | 33.63% | 31.466 |
| Latrodectus hesperus (Western black widow spider) | 17,364 | 31.17% | 2.02% | 197.44 | 30.44% | 28.896 |
| Eurytemora affinis (calanoid copepod) | 29,783 | 30.02% | 0.71% | 157.137 | 23.58% | 30.221 |
| All Pathways | KEGG Pathways | ||||
|---|---|---|---|---|---|
| Species | Proteins | Proteins Assigned to Pathways | Average Number of Proteins in Pathways | % Assigned to Pathways | Average Number of Proteins in Pathways |
| Apis mellifera (honeybee) | 15,314 | 29.27% | 3.41 | 17.57% | 20.23 |
| Drosophila melanogaster (fruit fly) | 30,724 | 37.73% | 8.77 | 21.24% | 49.08 |
| Tribolium castaneum (red flour beetle) | 18,534 | 30.03% | 4.22 | 16.99% | 23.68 |
| Varroa destructor (parasitic mite) | 30,221 | 41.55% | 9.63 | 23.50% | 54.62 |
| Athalia rosae (turnip sawfly) | 22,213 | 40.95% | 6.9 | 22.79% | 38.06 |
| Diaphorina citri (Asian citrus psyllid) | 19,049 | 40.07% | 5.88 | 23.72% | 34.75 |
| Limnephilus lunatus (caddisfly) | 13,292 | 38.09% | 3.92 | 22.94% | 23.10 |
| Cimex lectularius (Cimicidae bed bug) | 14,212 | 37.07% | 4.01 | 22.50% | 24.22 |
| Ceratitis capitata (Mediterranean fruit fly) | 12,318 | 35.91% | 3.35 | 21.36% | 19.78 |
| Oncopeltus fasciatus (large milkweed bug) | 19,793 | 32.51% | 4.9 | 18.36% | 27.53 |
| Agrilus planipennis (emerald ash borer) | 15,497 | 31.81% | 3.74 | 18.92% | 22.05 |
| Latrodectus hesperus (Western black widow spider) | 17,364 | 30.06% | 4.06 | 16.97% | 22.66 |
| Homalodisca vitripennis (glassy-winged sharpshooter) | 33,019 | 25.41% | 6.39 | 15.06% | 37.68 |
| Copidosoma floridanum (parasitoid wasp) | 19,869 | 25.35% | 3.83 | 14.43% | 21.56 |
| Eurytemora affinis (calanoid copepod) | 29,783 | 20.55% | 4.69 | 11.42% | 25.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Cooksey, A.M.; Childers, A.K.; Poelchau, M.F.; McCarthy, F.M. Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes. Insects 2021, 12, 748. https://doi.org/10.3390/insects12080748
Saha S, Cooksey AM, Childers AK, Poelchau MF, McCarthy FM. Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes. Insects. 2021; 12(8):748. https://doi.org/10.3390/insects12080748
Chicago/Turabian StyleSaha, Surya, Amanda M. Cooksey, Anna K. Childers, Monica F. Poelchau, and Fiona M. McCarthy. 2021. "Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes" Insects 12, no. 8: 748. https://doi.org/10.3390/insects12080748
APA StyleSaha, S., Cooksey, A. M., Childers, A. K., Poelchau, M. F., & McCarthy, F. M. (2021). Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes. Insects, 12(8), 748. https://doi.org/10.3390/insects12080748