
A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. DNA Sequencing and Assembly
2.3. Sequence Annotation and Analyses
2.4. Phylogenetic Analyses
2.5. Divergence Time Estimate
3. Results
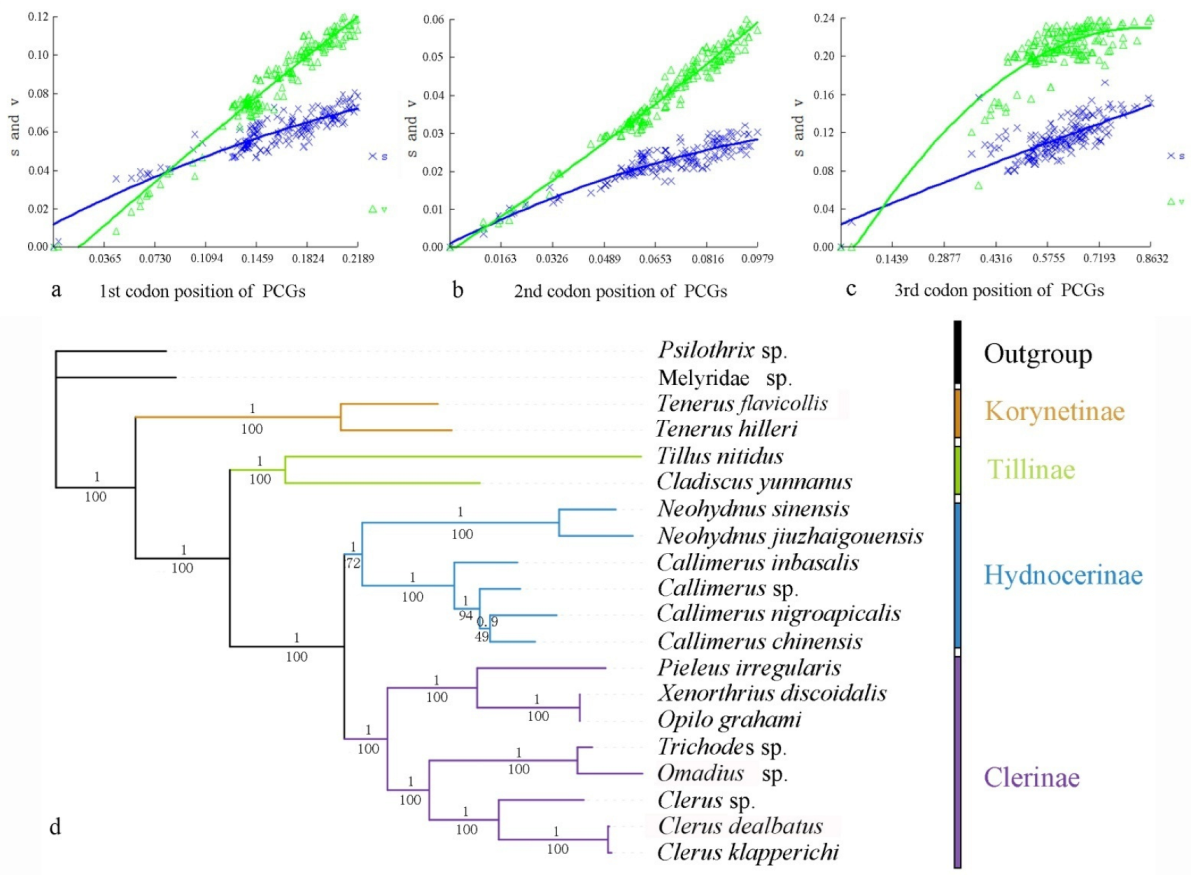
3.1. Phylogenetic Analyses
3.2. Divergence-Time Estimation
3.3. General Features of Mitochondrial Genome
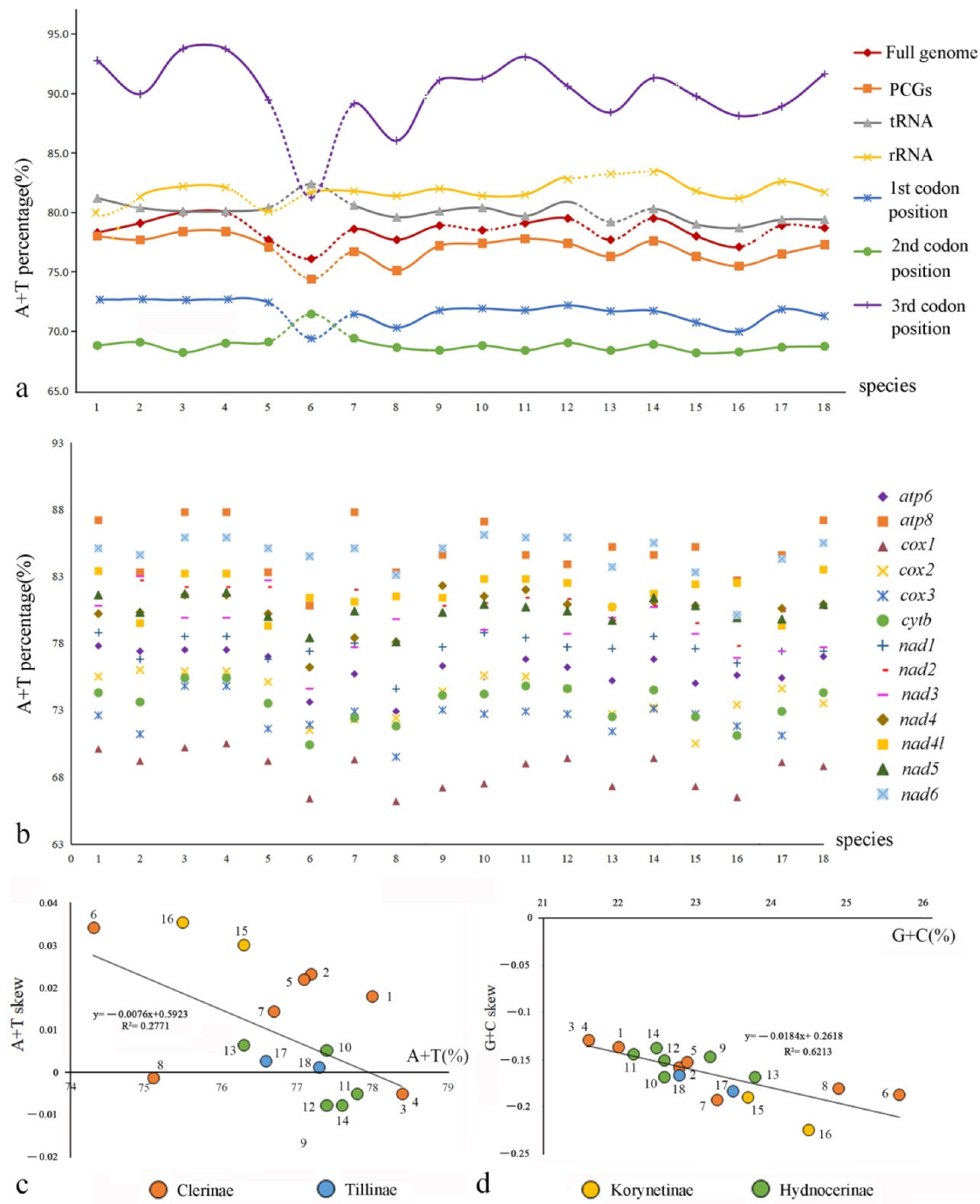
3.4. Nucleotide Composition
3.5. Codon Usage and Evolutionary Rates
4. Discussion
4.1. Phylogeny and Divergence-Time Estimation
4.2. The Characteristics of Mitochondrial Genomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Opitz, W. Classification, natural history, phylogeny, and subfamily composition of the Cleridae and generic content of the subfamilies (Coleoptera: Cleroidea). Entomol. Basiliensia Collect. Frey 2010, 32, 31–128. [Google Scholar] [CrossRef]
- Bocak, L.; Barton, C.; Crampton-Platt, A.; Chester, D.; Ahrens, D.; Vogler, A.P. Building the Coleoptera tree-of-life for >8000 species: Composition of public DNA data and fit with Linnaean classification. Syst. Entomol. 2014, 39, 97–110. [Google Scholar] [CrossRef]
- Gunter, N.L.; Leavengood, J.M.; Bartlett, J.S.; Chapman, E.G.; Cameron, S.L. A molecular phylogeny of the checkered beetles and a description of Epiclininae a new subfamily (Coleoptera: Cleroidea: Cleridae). Syst. Entomol. 2013, 38, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Gerstmeier, R. An overview of taxonomy and biology of the Cleridae (Coleoptera, Cleroidea, Cleridae). Gior. Ital. Entomol. 2014, 13, 481–494. [Google Scholar]
- Linneaus, C. Systema Naturae per Regna Tria Naturae, Secundum Classis, Ordines, Genera, Species cum Characteribus, Differentiis, Synonymis, Locis; Tomus, I., Editio, D.R., Eds.; Laurentius Salvius: Stockholm, Sweden, 1758; pp. 1–823. [Google Scholar] [CrossRef] [Green Version]
- Spinola, M. Monographie des Térédiles. Rev. Zool. 1841, 4, 70–76. [Google Scholar]
- Spinola, M. Essai Monographique sur les Clérites. Insectes Coléoptères; Imprimerie de Frères Ponthenier: Gênes, Italy, 1844. [Google Scholar]
- Lacordaire, T. Histoire Naturelle des Insectes. Genera des Coléoptères: Ou Exposé Méthodique et Critique de tous les Genres Proposés Jusque’ici Dans cet Ordre D’insectes; Libraire Encyclopédique de Roret: Paris, French, 1857; pp. 415–496. [Google Scholar]
- Schenkling, S. Coleoptera Malacodermata. Fam. Cleridae. In Genera Insectorum; Wytsman, P., Ed.; P. Wytsman: Bruxelles, Belgium, 1903; Volume 13, pp. 1–124. [Google Scholar]
- Schenkling, S. Coleopterorum Catalogus; W. Junk: Berlin, Germany, 1910; pp. 1–174. [Google Scholar]
- Gahan, C.J. Notes on Cleridae and descriptions of some new genera and species of this family of Coleoptera. Ann. Mag. Nat. Hist. 1910, 5, 55–76. [Google Scholar] [CrossRef]
- Chapin, E.A. Classification of the Philippine components of the Coleopterous family Cleridae. Philipp. J. Sci. 1924, 25, 159–286. [Google Scholar]
- Böving, A.G.; Craighead, F.C. An illustrated synopsis of the principal larval forms of the order Coleoptera. Entomol. Am. (NS) 1931, 11, 1–351. [Google Scholar]
- Crowson, R.A. A review of the classification of Cleroidea (Coleoptera), with descriptions of two new genera of Peltidae and of several new larval types. Trans. R. Entomol. Soc. Lond. 1964, 116, 275–327. [Google Scholar] [CrossRef]
- Winkler, J.R. A revision of the genus Dieropsis Gahan, 1908, type of a new subfamily Dieropsinae n. subf. (Coleoptera: Cleridae). Acta Univ. Carol. Biol. 1964, 1964, 305–329. [Google Scholar]
- Winkler, J.R. A revision of the subfamily Cleropiestinae subf. n. Acta Univ. Carol. Biol. 1980, 1978, 437–456. [Google Scholar]
- Winkler, J.R. Subfamilies and neutral terms proposed for groups of higher than subfamily in Cleridae (Coleoptera)—Purpose, definitions, identification key. Acta Univ. Carol. Biol. 1982, 1980, 517–531. [Google Scholar]
- Kolibáč, J. Revision of Thanerocleridae n. stat. (Coleoptera, Cleroidea). Mitt. Schweiz. Entomol. Ges. 1992, 65, 303–340. [Google Scholar]
- Kolibáč, J. Classification of the subfamilies of Cleridae (Coleoptera: Cleroidea). Acta Musei Morav. Sci. Nat. 1997, 81, 307–361. [Google Scholar]
- Leschen, R.A.B. 9. Cleroidea Latreille, 1802, Introduction and Phylogeny. In Coleoptera, Beetles: Morphology and Systematics (Elateroidea, Bostrichiformia, Cucujiformia Partim); Leschen, R.B., Beutel, R.G., Lawrence, J.F., Eds.; Walter de Gruyter: Berlin, German, 2010; Volume 2, pp. 237–280. [Google Scholar]
- Opitz, W. Classification, natural history, and evolution of Neorthopleurinae subfam.nov. (Coleoptera, Cleridae) Part I. Generic composition of the subfamily and key to genera. Entomol. Basil. Coll. Frey. 2009, 31, 135–207. [Google Scholar]
- Gimmel, M.L.; Bocakova, M.; Gunter, N.L.; Leschen, R.A.B. Comprehensive phylogeny of the Cleroidea (Coleoptera: Cucujiformia). Syst. Entomol. 2019, 44, 527–558. [Google Scholar] [CrossRef]
- Bartlett, J.S. A Preliminary Suprageneric Classification for Clerinae (Coleoptera: Cleridae) based on Molecular and Morphological Evidence, Including a Review of Tegminal Terminology. Ann. Zool. 2021, 71, 737–766. [Google Scholar] [CrossRef]
- Kolibáč, J.; Huang, D. The oldest known clerid fossils from the Middle Jurassic of China, with a review of Cleridae systematics (Coleoptera). Syst. Entomol. 2016, 41, 808–823. [Google Scholar] [CrossRef]
- Kolibáč, J.; Bocakova, M.; Liebherr, J.K.; Ramage, T.; Porch, N. Extinct and extant Pacific Trogossitidae and the evolution of Cleroidea (Coleoptera) after the Late Triassic biotic crisis. Zool. J. Linn. Soc. 2021, 191, 846–882. [Google Scholar] [CrossRef]
- Bocakova, M.; Constantin, R.; Bocak, L. Molecular phylogenetics of the melyridlineage (Coleoptera: Cleroidea). Cladistics 2012, 28, 117–129. [Google Scholar] [CrossRef]
- Bocakova, M.; Bocak, L.; Gimmel, M.L.; Motyka, M.; Vogler, A.P. Aposematism and mimicry in soft-bodied beetles of the superfamily Cleroidea (Insecta). Zool. Scr. 2016, 45, 9–21. [Google Scholar] [CrossRef]
- Hunt, T.; Bergsten, J.; Levkanicova, Z.; Papadopoulou, A.; John, O.S.; Wild, R.; Hammond, P.M.; Ahrens, D.; Balke, M.; Caterino, M.S. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- McKenna, D.D.; Wild, A.L.; Kanda, K.; Bellamy, C.L.; Beutel, R.G.; Caterino, M.S.; Farnum, C.W.; Hawks, D.C.; Ivie, M.A.; Jameson, M.L.; et al. Beetle tree of life reveals that Coleoptera survived end-Permian mass extinction to diversify during the Cretaceous terrestrial revolution. Syst. Entomol. 2015, 40, 835–880. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, E.F.A.; Seidel, M.; Arriaga-Varela, E.; Hájek, J.I.; Kral, D.; Sekerka, L.U.; Short, A.E.; Fikáček, M.A. The peril of dating beetles. Syst. Entomol. 2017, 42, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Q.; Che, L.H.; Li, Y.; Liang, D.; Pang, H.; Ślipiński, A.; Zhang, P. Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Commun. 2018, 9, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, D.K.; Friberg., U.; Lindell., J. Evolutionary implications of non-neutral mitochondrial genetic variation. Trends. Ecol. Evol. 2008, 23, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. A mitochondrial genome phylogeny of Diptera: Whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Li, H.; Liu, G.H.; Wang, W.; James, P.; Colwell, D.D.; Tran, A.; Gong, S.; Cai, W.; Shao, R. Mitochondrial genome fragmentation unites the parasitic lice of Eutherian mammals. Syst. Biol. 2019, 68, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Saccone, C.; Gissi, C.; Lanave, C.; Larizza, A.; Pesole, G.; Reyes, A. Evolution of the mitochondrial genetic system: An overview. Gene 2000, 261, 153–159. [Google Scholar] [CrossRef]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Song, F.; Zhao, Y.; Wilson, J.J.; Cai, W. Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Wang, Q.; Li, K.; Pape, T.; Zhang, D. Molecular and morphological characterization of third instar Palaearctic horse stomach bot fly larvae (Oestridae: Gasterophilinae, Gasterophilus). Vet. Parasitol. 2018, 262, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shao, R.; Song, N.; Song, F.; Jiang, P.; Li, Z.; Cai, W. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Zhang, Q.; Zhang, L.; Guo, Z.; Liu, Y.; Shen, Y.; Shao, R. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 2016, 104, 99–111. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of holometabola with mitochondrial genome data and Bayesian siteheterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Castro, L.R.; Austin, A.D.; Dowton, M. Contrasting rates of mitochondrial molecular evolution in parasitic Diptera and Hymenoptera. Mol. Biol. Evol. 2002, 19, 1100–1113. [Google Scholar] [CrossRef]
- Oliveira, D.C.S.G.; Raychoudhury, R.; Lavrov, D.V.; Werren, J.H. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae). Mol. Biol. Evol. 2008, 25, 2167–2180. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Yang, G.Y. Study on Systematics of Cleridae from China (Coleoptera); Institute of Zoology, Chinese Academy of Sciences: Beijing, China, 2012. [Google Scholar]
- Zhou, X.; Li, Y.; Liu, S.; Yang, Q.; Su, X.; Zhou, L.; Tang, M.; Fu, R.; Li, J.; Huang, Q. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. GigaScience 2013, 2, 2047-217X. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single--cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Gärtner, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSPv5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Bowker, A.H. A test for symmetry in contingency tables. J. Am. Stat. Assoc. 1948, 43, 572–574. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Ge, X.; Xie, G.; Liu, H.; Yang, Y. First Complete Mitochondrial Genome of Melyridae (Coleoptera, Cleroidea): Genome Description and Phylogenetic Implications. Insects 2021, 12, 87. [Google Scholar] [CrossRef]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-Level Sampling of Mitochondrial Genomes in Coleoptera: Compositional Heterogeneity and Phyloge-netics. Genome Biol. Evol. 2016, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. Mafft Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usa-bility. Mol. Biol. Evol. 2013, 4, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Arndt, V.H.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBaye 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Cockerell, T.D.A. The fauna of the Sunchal (or Margas Verdes) formation, Northern Argentina. Am. Mus. Novit. 1936, 886, 1–9. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4.3. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 16 January 2022).
- Hurst, L.D.; Merchant, A.R. High guanine-cytosine content is not an adaptation to high temperature: A comparative analysis amongst prokaryotes. Proc. Biol. Sci. 2001, 268, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L.; Lo, N.; Bourguignon, T.; Svenson, G.J.; Evans, T.A. A Mitochondrial Genome Phylogeny of Termites (Blattodea: Termitoidae): Robust Support for Interfamilial Relationships and Molecular Synapomorphies Define Major Clades. Mol. Phylogenet. Evol. 2012, 65, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Su, T.J.; Chesters, D.; Wang, S.D.; Ho, S.Y.W.; Zhu, C.S.; Chen, X.L.; Zhang, C.T. The mitochondrial genome of Elodia flavipalpis Aldrich (Diptera: Tachinidae) and the evolutionary timescale of tachinid flies. PLoS ONE 2013, 8, e61814. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Ribera, I.; Bertranpetit, J.; Balke, M. Nucleotide substitution rates for the full set of mitochondrial p rotein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 2010, 56, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 2006, 38, 100–116. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Tomita, M.; Arakawa, K. Accelerated laboratory evolution reveals the influence of replication on the GC skew in Escherichia coli. Genome Biol. Evol. 2018, 10, 3110–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.H.; Lu, G.; Bork, P.; Hu, S.; Lercher, M.J. Energy efficiency trade-offs drive nucleotide usage in transcribed regions. Nat. Commun. 2016, 7, 11334. [Google Scholar] [CrossRef] [Green Version]
- McFerrin, L.G.; Stone, E.A. The non-random clustering of non-synonymous substitutions and its relationship to evolutionary rate. BMC Genom. 2011, 12, 415. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.Z.; Yan, H.B.; Guo, A.J.; Zhu, X.Q.; Wang, Y.C.; Shi, W.G.; Chen, H.T.; Zhan, F.; Zhang, S.H. Complete mitochondrial genomes of Taeniamulticeps, T. hydatigena and T. pisiformis: Additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genom. 2010, 11, 447. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Easy, R.H.; King, S.D.; Cone, D.K.; You, P. Comparative analyses within Gyrodactylus (Platyhelminthes: Monogenea) mitochondrial genomes and conserved polymerase chain reaction primers for gyrodactylid mitochondrial DNA. J. Fish Dis. 2017, 40, 541–555. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Z.; Niu, L.; Wang, Q.; Wang, C.; Lan, J.; Deng, J.; Fu, Y.; Nie, H.; Yan, N. The mitochondrial genome of Baylisascaris procyonis. PLoS ONE 2011, 6, e27066. [Google Scholar] [CrossRef] [Green Version]
- Pureum, N.; Wook, J.K.; Jun-Ho, S.; Inkyu, P.; Goya, C.; Byeong, C.M. Rapid and Simple Species Identification of Cicada Exuviae Using COI-Based SCAR Assay. Insects 2020, 11, 168. [Google Scholar] [CrossRef] [Green Version]
- Elaine, F.; Alexandre, R.Z.; Paulo, C.R.; João, P.; Naldi, S.; Rute, B.; Benjamin, P.O.; Maria, C.A. Conserved Numts Mask a Highly Divergent Mitochondrial-COI Gene in a Species Complex of Australian Stingless Bees Tetragonula (Hymenoptera: Apidae). Mitochondrial. DNA Part A 2019, 30, 806–817. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species ID | Family/ Subfamily | Species | Depository/ Voucher No. | Locality/ Collection Information | Geographic Coordinates | GenBank No. |
|---|---|---|---|---|---|---|
| 1 | Cleridae/ Clerinae | Clerus sp. | MHBU, 2CA0214 | China, Beijing: Mentougou, Xiaolongmen, V-12-2018 | 115°27′4.22″ E, 39°58′36.87″ N | MZ464014 |
| 2 | Clerus dealbatus | MHBU, CAN0129 | China, Shaanxi: Yangxian, Youdeng, VI-24-2017 | 107°22′30.72″ E, 33°27′7.80″ N | MZ490582 | |
| 3 | Xenorthrius discoidalis | MHBU, CAN0019 | China, Gansu: Tianshui, Maiji, Dangchuan, Fangmatan, VIII-9-2018 | 106°6′48.37″ E, 34°25′3.38″ N | MZ490583 | |
| 4 | Opilo grahami | MHBU, CAN0176 | China, Yunnan: Jingdong, Ailaoshan, Xujiaba, VIII-17-2013 | 101°1′14.74″ E, 24°31′15.90″ N | MZ488575 | |
| 5 | Clerus klapperichi | MHBU, 2CA0167 | China, Zhejiang: Tianmushan, Xianrending, V-6-2018 | 119°26′43.64″ E, 30°20′43.87″ N | MZ475053 | |
| 6 | Omadius sp. | MHBU, 2CA0218 | China, Xizang: Nyingchi, Medôg, VIII-14-2016, | 95°20′30.47″ E, 29°19′55.29″ N | MZ490580 | |
| 7 | Trichodes sp. | MHBU, CAN0128 | China, Shaanxi: Yangxian, Youdengvill., VI-24-2017 | 107°22′30.95″ E, 33°27′7.72″ N | MZ490584 | |
| 8 | Pieleus irregularis | MHBU, 2CA0165 | China, Zhejiang: Tianmushan, Xianrending, V-6-2018 | 119°26′43.64″ E, 30°20′43.87″ N | MZ488576 | |
| 9 | Cleridae/ Hydnocerinae | Callimerus chinensis | MHBU, CAN0173 | China, Yunnan: Lancang, Donghe, Shangbanggan, XI-18-2017 | 100°04′06.01″ E, 55°44′09″ N | MZ464016 |
| 10 | Callimerus inbasalis | MHBU, 2CA0173 | China, Yunnan: Puer, Lancang, VII-5-2017 | 99°56′2.49″ E, 22°33′33.22″ N | MZ464017 | |
| 11 | Callimerus sp. | MHBU, 2CA0166 | China, Hunan: Shaoyang, Chengbu, Dankou, Taiping, V-6-2018 | 110°14′52.64″ E, 26°21′25.14″ N | MZ488577 | |
| 12 | Callimerus nigroapicalis | MHBU, CAN0183 | China, Hainan: Ledong, Jiangfengling, IV-10-2019 | 108°54′32.99″ E, 18°43′49.69″ N | MZ475052 | |
| 13 | Neohydnus sinensis | MHBU, 2CA0035 | China, Guangxi: Wuming, Damingshan, V-21-2011 | 108°20′33.57″ E, 23°31′45.78″ N | MZ464019 | |
| 14 | Neohydnus jiuzhaigouensis | MHBU, CAN0226 | China, Hubei: Shennongjia, Tiechanghe, VI-25-2019 | 110°46′15.17″ E, 31°39′51.54″ N | MZ464018 | |
| 15 | Cleridae/ Korynetinae | Tenerus flavicollis | MHBU, 2CA0146 | China, Yunnan: IV-29-2010 | 102°55′40.02″ E, 25°0′3.18″ N | MZ488578 |
| 16 | Tenerus hilleri | MHBU, 2CA0172 | China, Sichuan: Pengzhou, Danjingshan, VI-7-2019, | 103°50′21.24″ E, 31°5′9.99″ N | MZ488579 | |
| 17 | Cleridae/ Tillinae | Tillus nitidus | MHBU, 2CA0216 | China, Shaanxi: Zhouzhi, Louguantai, VI-25-2008 | 108°19′58.28″ E, 34°3′49.19″ N | MZ490581 |
| 18 | Cladiscus yunnanus | MHBU, 2CA0079 | China, Yunnan: Xishuangbanna, Tropical Botanical Garden, VI-2-2015 | 101°16′44.26″ E, 21°55′20.83″ N | MZ464015 | |
| Out- group | Melyridae/ Dasytinae | Dasytinae sp. Psilothrix sp. | JX412765 JX412801 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Liu, H.; Ge, X.; Yang, G.; Xie, G.; Yang, Y. A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea). Insects 2022, 13, 118. https://doi.org/10.3390/insects13020118
Yuan L, Liu H, Ge X, Yang G, Xie G, Yang Y. A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea). Insects. 2022; 13(2):118. https://doi.org/10.3390/insects13020118
Chicago/Turabian StyleYuan, Lilan, Haoyu Liu, Xueying Ge, Ganyan Yang, Guanglin Xie, and Yuxia Yang. 2022. "A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea)" Insects 13, no. 2: 118. https://doi.org/10.3390/insects13020118
APA StyleYuan, L., Liu, H., Ge, X., Yang, G., Xie, G., & Yang, Y. (2022). A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea). Insects, 13(2), 118. https://doi.org/10.3390/insects13020118