

Chromosome-Level Genome Assembly of Papilio elwesi Leech, 1889 (Lepidoptera: Papilionidae)


{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Genome Survey and Assembly
2.3. Genome Annotation
2.4. Gene Family Identification and Evolution
2.5. Chromosomal Synteny
3. Results and Discussion
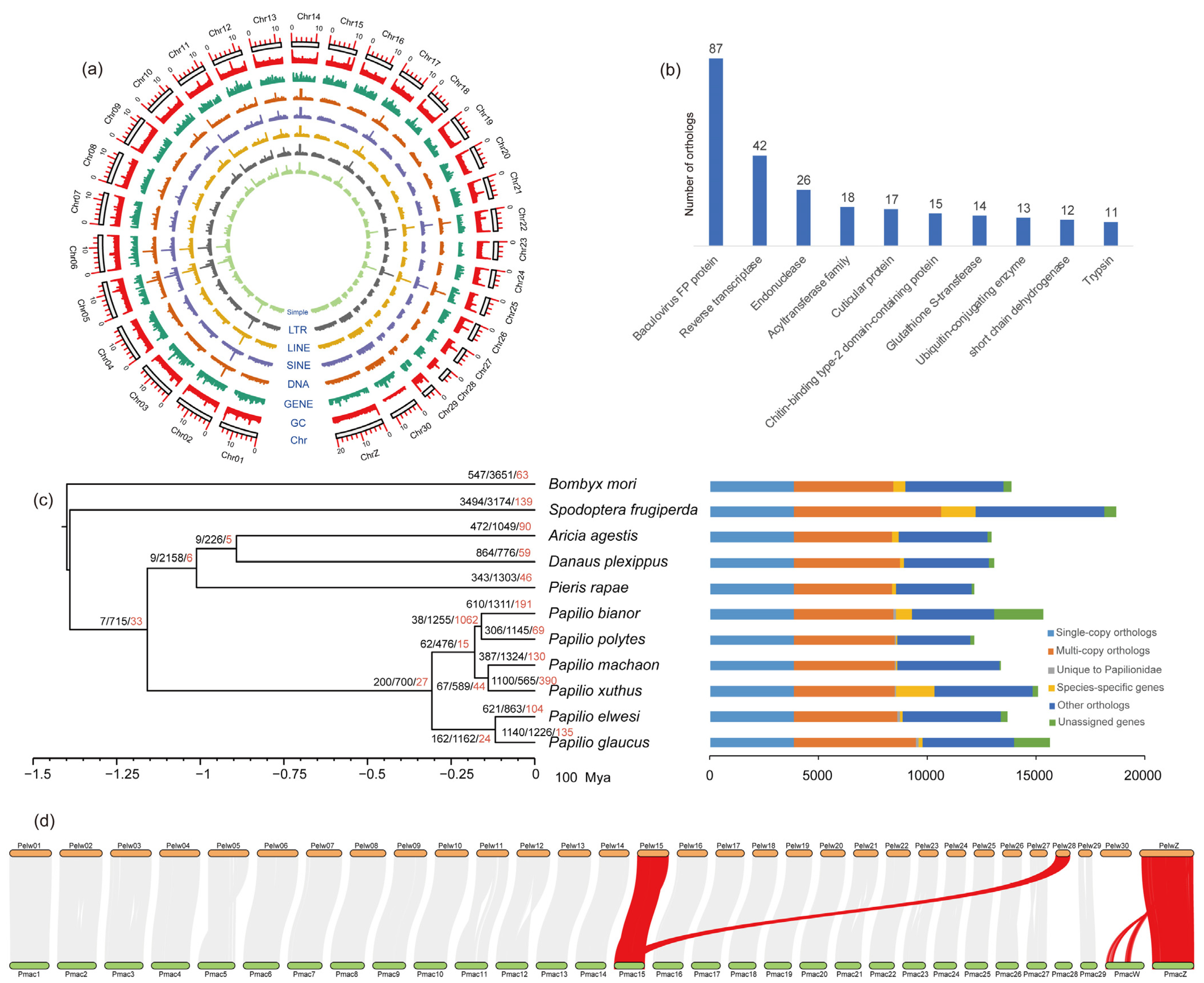
3.1. Sequencing and Genome Survey
3.2. Genome and Mitochondrion Assembly
3.3. Genome Annotation
3.4. Gene Family Evolution and Phylogenetic Relationships
3.5. Chromosomal Synteny
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Nieukerken, E.J.; Kaila, L.; Kitching, I.J.; Kristensen, N.P.; Lees, D.C.; Minet, J.; Mitter, C.; Mutanen, M.; Regier, J.C.; Simonsen, T.J.; et al. Order Lepidoptera Linnaeus, 1758. Zootaxa 2011, 3148, 212–221. [Google Scholar] [CrossRef]
- Joron, M.; Mallet, J.L.B. Diversity in mimicry: Paradox or paradigm? Trends Ecol. Evol. 1998, 13, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Brakefield, P.M.; French, V. Butterfly Wings: The evolution of eevelopment of colour patterns. BioEssays 1999, 21, 391–401. [Google Scholar] [CrossRef]
- Kunte, K. The diversity and evolution of Batesian mimicry in Papilio swallowtail butterflies. Evolution 2009, 63, 2707–2716. [Google Scholar] [CrossRef]
- McMillan, W.O.; Monteiro, A.; Kapan, D.D. Development and evolution on the wing. Trends Ecol. Evol. 2002, 17, 125–133. [Google Scholar] [CrossRef]
- Beldade, P.; Brakefield, P.M. The genetics and evo–devo of butterfly wing patterns. Nat. Rev. Genet. 2002, 3, 442–452. [Google Scholar] [CrossRef]
- Espeland, M.; Breinholt, J.; Willmott, K.R.; Warren, A.D.; Vila, R.; Toussaint, E.; Maunsell, S.C.; Aduse-Poku, K.; Talavera, G.; Eastwood, R. A Comprehensive and Dated Phylogenomic Analysis of Butterflies. Curr. Biol. 2018, 28, 770–778.e5. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.M.; Morris, M.G. Threatened swallowtail butterflies of the world. In The IUCN Red Data Book; IUCN: Cambridge, UK, 1985. [Google Scholar]
- Scriber, J.M.; Tsubaki, Y.; Lederhouse, R.C. Swallowtail Butterflies: Their Ecology and Evolutionary Biology; Scientific Publishers: Gainesville, FL, USA, 1995. [Google Scholar]
- Heikkila, M.; Kaila, L.; Mutanen, M.; Pena, C.; Wahlberg, N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. Biol. Sci. 2012, 279, 1093–1099. [Google Scholar] [CrossRef]
- Kawahara, A.Y.; Breinholt, J.W. Phylogenomics provides strong evidence for relationships of butterflies and moths. Proc. R. Soc. B 2014, 281, 20140970. [Google Scholar] [CrossRef] [Green Version]
- Mitter, C.; Davis, D.R.; Cummings, M.P. Phylogeny and Evolution of Lepidoptera. Annu. Rev. Èntomol. 2017, 62, 265–283. [Google Scholar] [CrossRef]
- Li, X.; Fan, D.; Zhang, W.; Liu, G.; Zhang, L.; Zhao, L.; Fang, X.; Chen, L.; Dong, Y.; Chen, Y.; et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 2015, 6, 8212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markert, M.J.; Zhang, Y.; Enuameh, M.S.; Reppert, S.M.; Wolfe, S.A.; Merlin, C. Genomic Access to Monarch Migration Using TALEN and CRISPR/Cas9-Mediated Targeted Mutagenesis. G3 Genes|Genomes|Genetics 2016, 6, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Reed, R.D. Genome editing in butterflies reveals that spalt promotes and Distal-less represses eyespot colour patterns. Nat. Commun. 2016, 7, 11769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Yang, J.; Dai, X.; Xie, F.; He, J.; Dong, Z.; Mao, J.; Liu, G.; Chang, Z.; Zhao, R.; et al. Chromosomal-level reference genome of Chinese peacock butterfly (Papilio bianor) based on third-generation DNA sequencing and Hi-C analysis. Gigascience 2019, 8, giz128. [Google Scholar] [CrossRef]
- Tunstrom, K.; Wheat, C.W.; Parmesan, C.; Singer, M.C.; Mikheyev, A.S. A genome for Edith’s checkerspot butterfly: An insect with complex host-adaptive suites and rapid evolutionary responses to environmental changes. Genome Biol. Evol. 2022, 14, evac113. [Google Scholar] [CrossRef]
- Guiglielmoni, N.; Houtain, A.; Derzelle, A.; Van Doninck, K.; Flot, J.-F. Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinform. 2021, 22, 1–23. [Google Scholar] [CrossRef]
- Ellis, E.A.; Storer, C.G.; Kawahara, A.Y. De novo genome assemblies of butterflies. Gigascience 2021, 10, giab041. [Google Scholar] [CrossRef]
- Challi, R.J.; Kumar, S.; Dasmahapatra, K.K.; Jiggins, C.D.; Blaxter, M. Lepbase: The lepidopteran genome database. bioRxiv 2016. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, C.R.; Selegue, J.E.; Monteiro, A.; French, V.; Brakefield, P.M.; Carroll, S.B. The generation and diversification of butterfly eyespot color patterns. Curr. Biol. 2001, 11, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Loehlin, D.W.; Carroll, S.B. Sex, lies and butterflies. Nature 2014, 507, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mazo-Vargas, A.; Reed, R.D. Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. Proc. Natl. Acad. Sci. USA 2017, 114, 10707–10712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahara, K.; Yoshido, A.; Traut, W. Sex chromosome evolution in moths and butterflies. Chromosom. Res. 2011, 20, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalíková, M.; Zrzavá, M.; Hladová, I.; Nguyen, P.; Šonský, I.; Flegrová, M.; Kubíčková, S.; Voleníková, A.; Kawahara, A.Y.; Peters, R.S.; et al. New Insights into the Evolution of the W Chromosome in Lepidoptera. J. Hered. 2017, 108, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraïsse, C.; Picard, M.A.L.; Vicoso, B. The deep conservation of the Lepidoptera Z chromosome suggests a non-canonical origin of the W. Nat. Commun. 2017, 8, 1486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ding, Y.; Zhou, Q.-S.; Wu, J.; Luo, A.; Zhu, C.-D. A High-quality Draft Genome Assembly of Sinella curviseta: A Soil Model Organism (Collembola). Genome Biol. Evol. 2019, 11, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bushnell, B. BBtools. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022).
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F.A.; Hubley, R.; Green, P. 2013–2015. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022).
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 1–14. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keilwagen, J.; Hartung, F.; Paulini, M.; Twardziok, S.O.; Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018, 19, 189. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2008, 37, D380–D386. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Steenwyk, J.L.; Buida, T.J.; Labella, A.L.; Li, Y.; Shen, X.-X.; Rokas, A. PhyKIT: A broadly applicable UNIX shell toolkit for processing and analyzing phylogenomic data. Bioinformatics 2021, 37, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Han, M.V.; Thomas, G.W.; Lugo-Martinez, J.; Hahn, M.W. Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Bian, D.; Ye, W.; Dai, M.; Lu, Z.; Li, M.; Fang, Y.; Qu, J.; Su, W.; Li, F.; Sun, H.; et al. Phylogenetic relationships of Limacodidae and insights into the higher phylogeny of Lepidoptera. Int. J. Biol. Macromol. 2020, 159, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Liu, Y.; Zheng, X.; Zhang, R.; Feng, K.; Yue, B.; Du, C.; Zhou, C. Characterization of Seventeen Complete Mitochondrial Genomes: Structural Features and Phylogenetic Implications of the Lepidopteran Insects. Insects 2022, 13, 998. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Èntomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Allio, R.; Scornavacca, C.; Nabholz, B.; Clamens, A.-L.; Sperling, F.A.; Condamine, F.L. Whole Genome Shotgun Phylogenomics Resolves the Pattern and Timing of Swallowtail Butterfly Evolution. Syst. Biol. 2020, 69, 38–60. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, H.; Chen, C.; Hao, D. Tolerance to dietary linalool primarily involves co-expression of cytochrome P450s and cuticular proteins in Pagiophloeus tsushimanus (Coleoptera: Curculionidae) larvae using SMRT sequencing and RNA-seq. BMC Genom. 2023, 24, 34. [Google Scholar] [CrossRef]
- Dai, W.; Mank, J.E.; Ban, L. Repeated origin of the W chromosome from the Z chromosome in Lepidoptera. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yoshido, A.; Marec, F.; Sahara, K. The fate of W chromosomes in hybrids between wild silkmoths, Samia cynthia ssp.: No role in sex determination and reproduction. Heredity 2016, 116, 424–433. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Z.; Ding, Y.; Zhang, S.; Li, L.; Ma, F. Chromosome-Level Genome Assembly of Papilio elwesi Leech, 1889 (Lepidoptera: Papilionidae). Insects 2023, 14, 304. https://doi.org/10.3390/insects14030304
Pan Z, Ding Y, Zhang S, Li L, Ma F. Chromosome-Level Genome Assembly of Papilio elwesi Leech, 1889 (Lepidoptera: Papilionidae). Insects. 2023; 14(3):304. https://doi.org/10.3390/insects14030304
Chicago/Turabian StylePan, Zhixiang, Yinhuan Ding, Shusheng Zhang, Luxian Li, and Fangzhou Ma. 2023. "Chromosome-Level Genome Assembly of Papilio elwesi Leech, 1889 (Lepidoptera: Papilionidae)" Insects 14, no. 3: 304. https://doi.org/10.3390/insects14030304
APA StylePan, Z., Ding, Y., Zhang, S., Li, L., & Ma, F. (2023). Chromosome-Level Genome Assembly of Papilio elwesi Leech, 1889 (Lepidoptera: Papilionidae). Insects, 14(3), 304. https://doi.org/10.3390/insects14030304