
Integrated Omics Analysis Reveals Key Pathways in Cotton Defense against Mirid Bug (Adelphocoris suturalis Jakovlev) Feeding


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Insect Infestation
2.2. Metabolomic Sequencing
2.3. iTRAQ Sequencing
2.4. Quantitative Real-Time PCR (qRT-PCR)
2.5. Data Analysis
3. Results
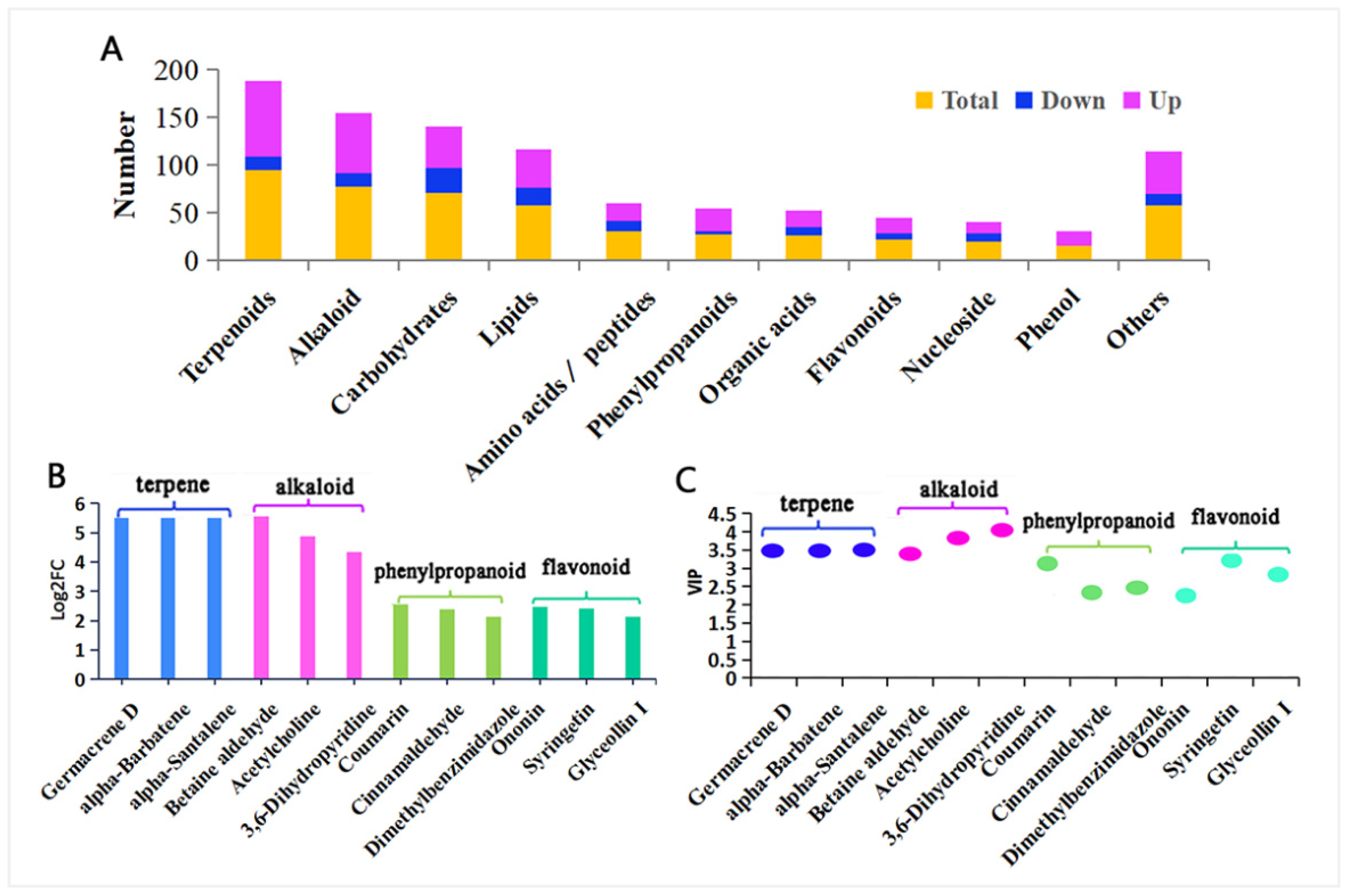
3.1. Metabolite Identification
3.1.1. Identification of Differential Metabolites
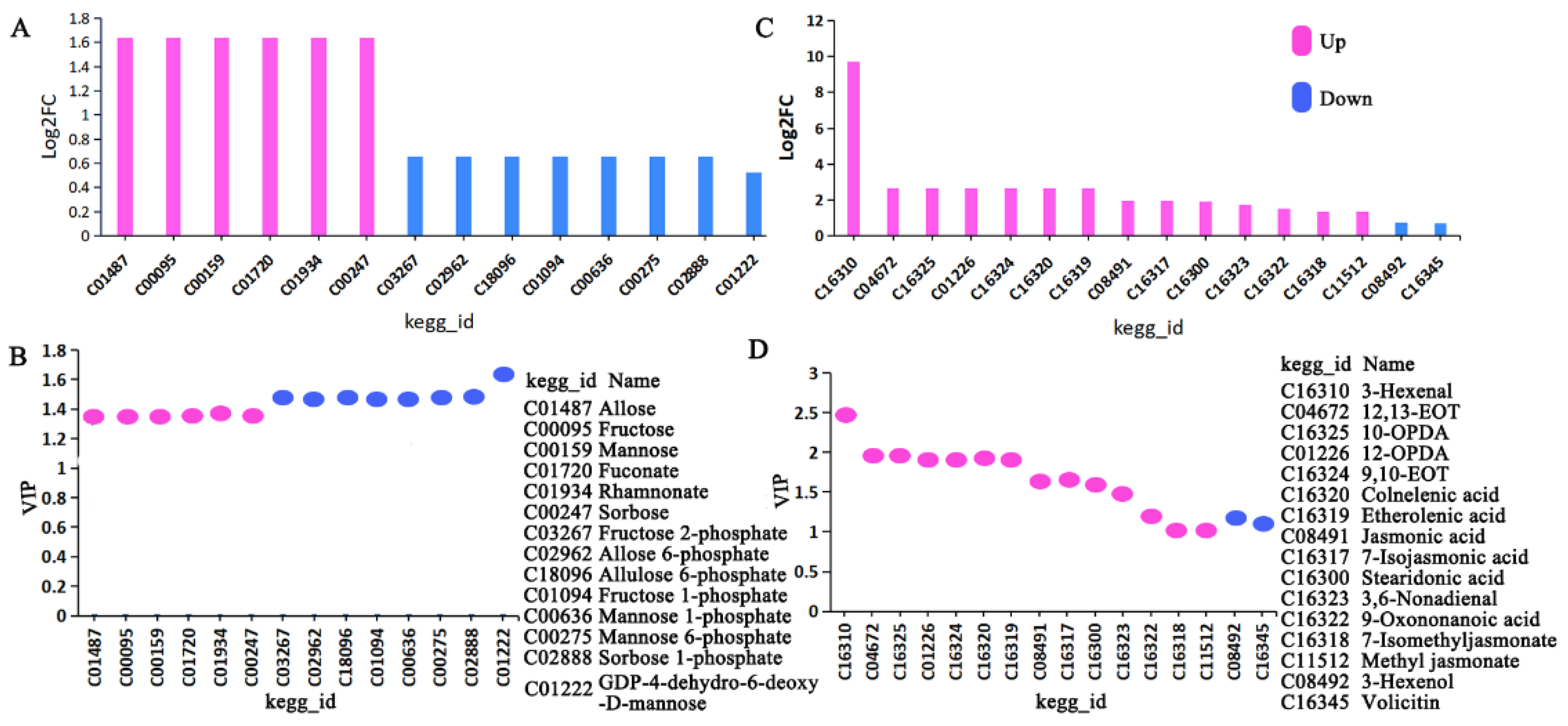
3.1.2. KEGG Annotation and Functional Analysis of Differential Metabolites
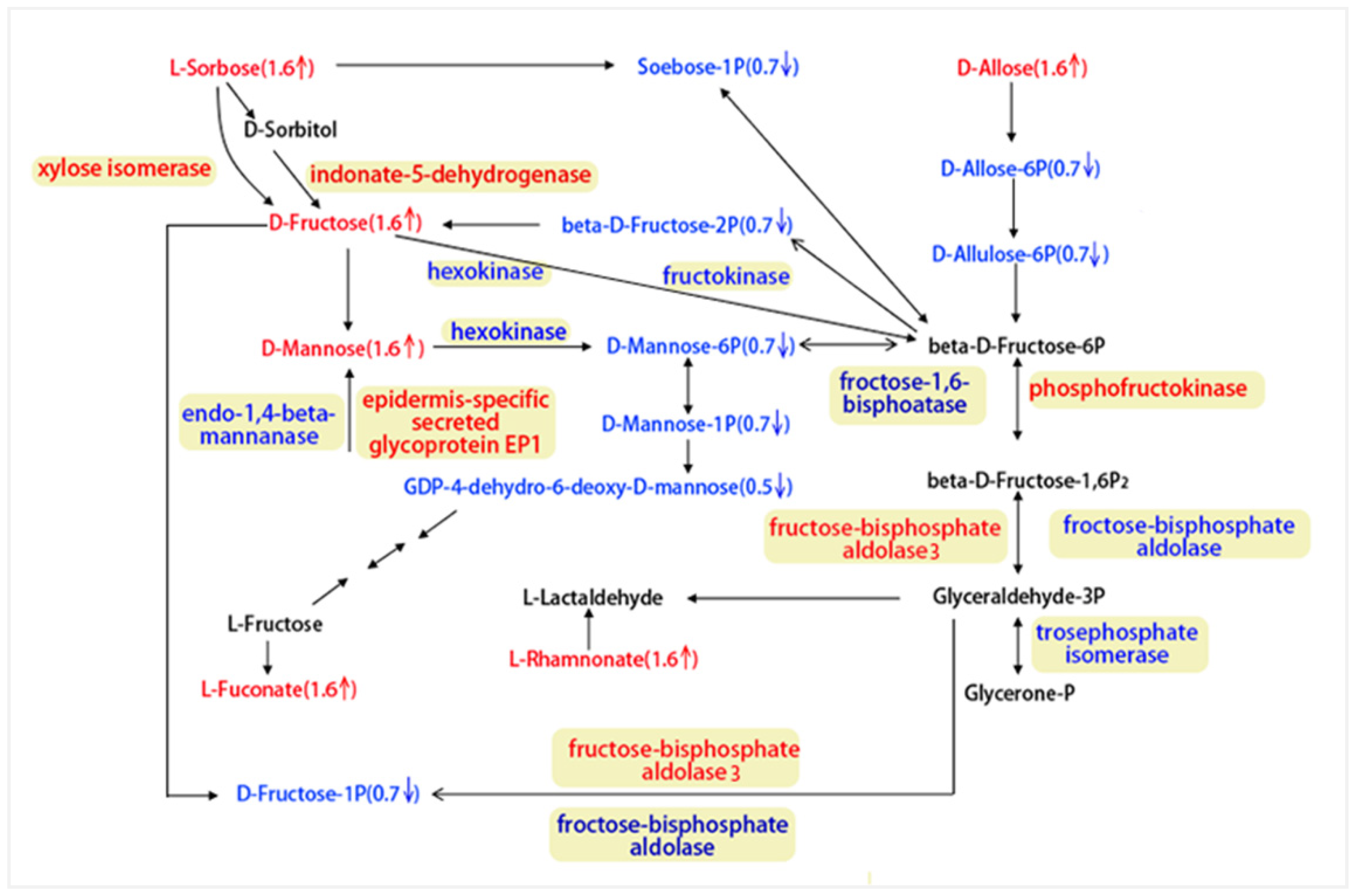
3.1.3. The Two Key Metabolic Pathways
3.2. iTRAQ Identification of Proteins
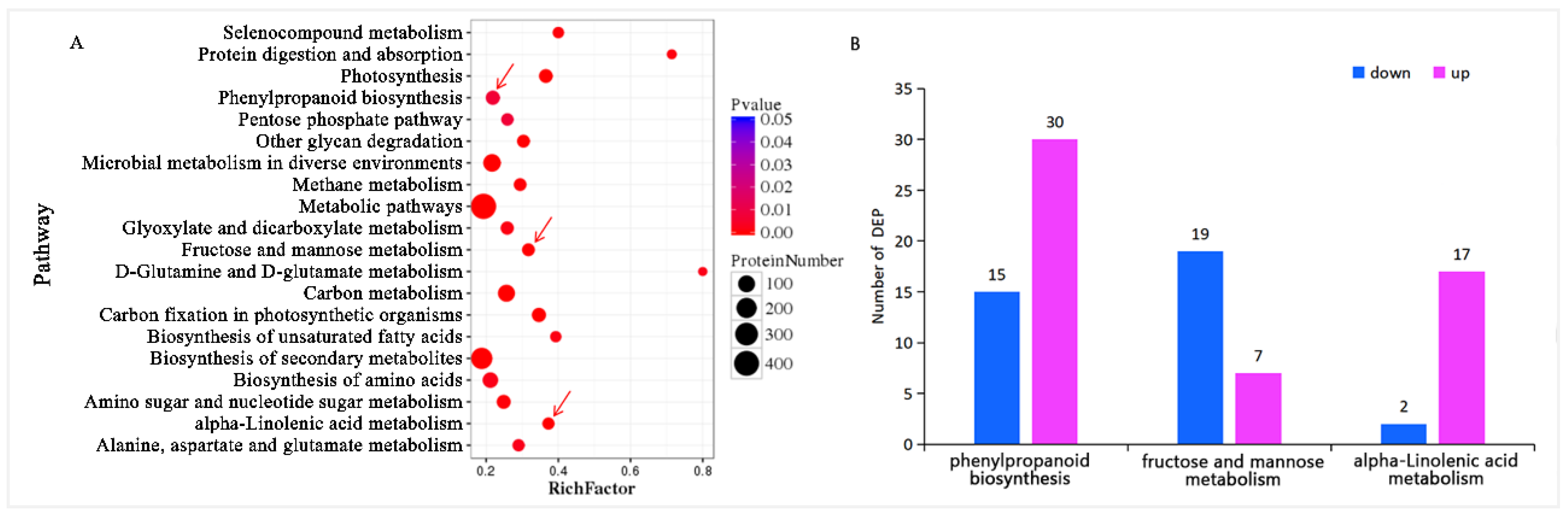
3.2.1. Functional Categories of Differentially Expressed Proteins
3.2.2. Pathway Enrichment of Differential Proteins
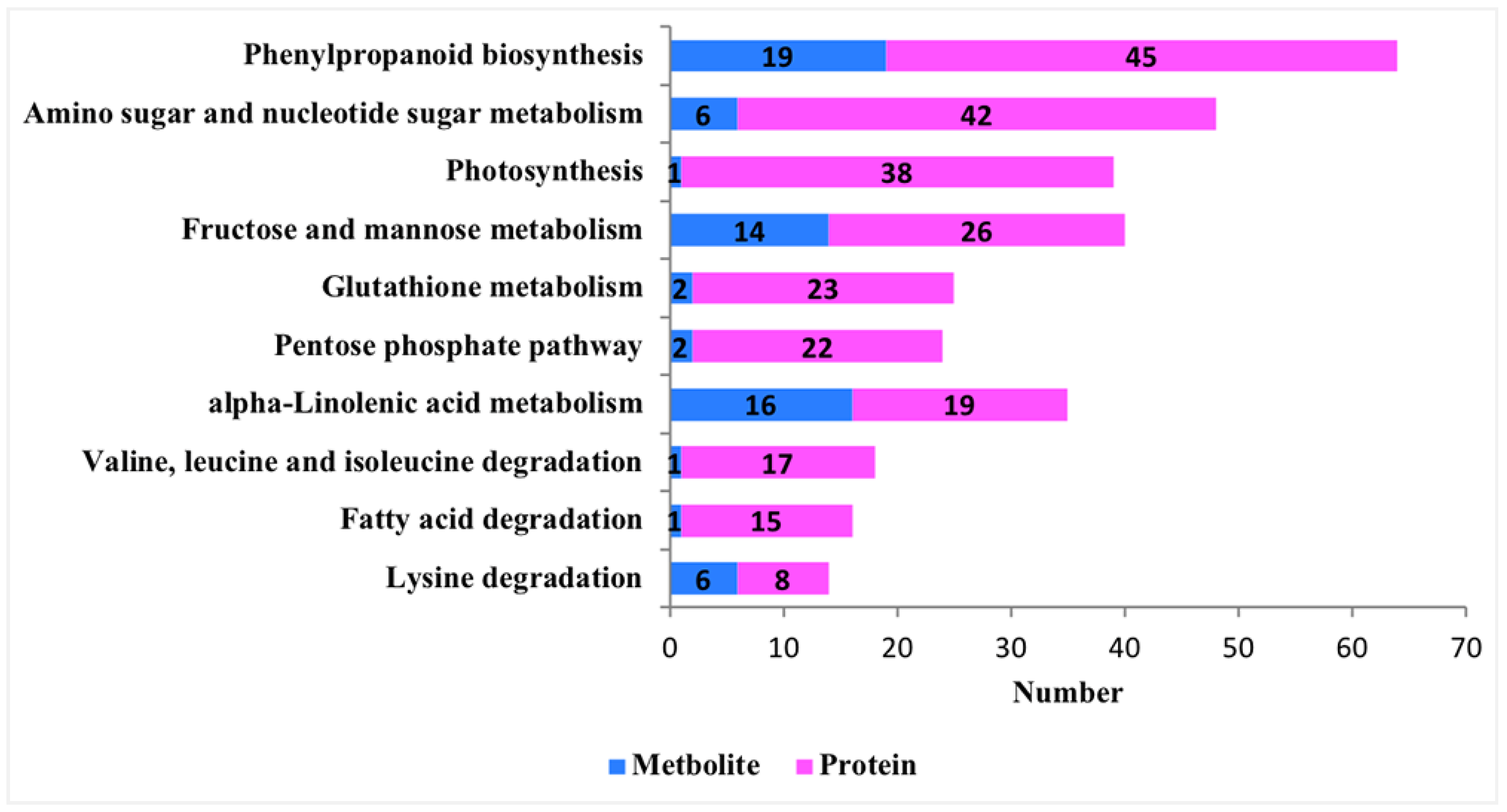
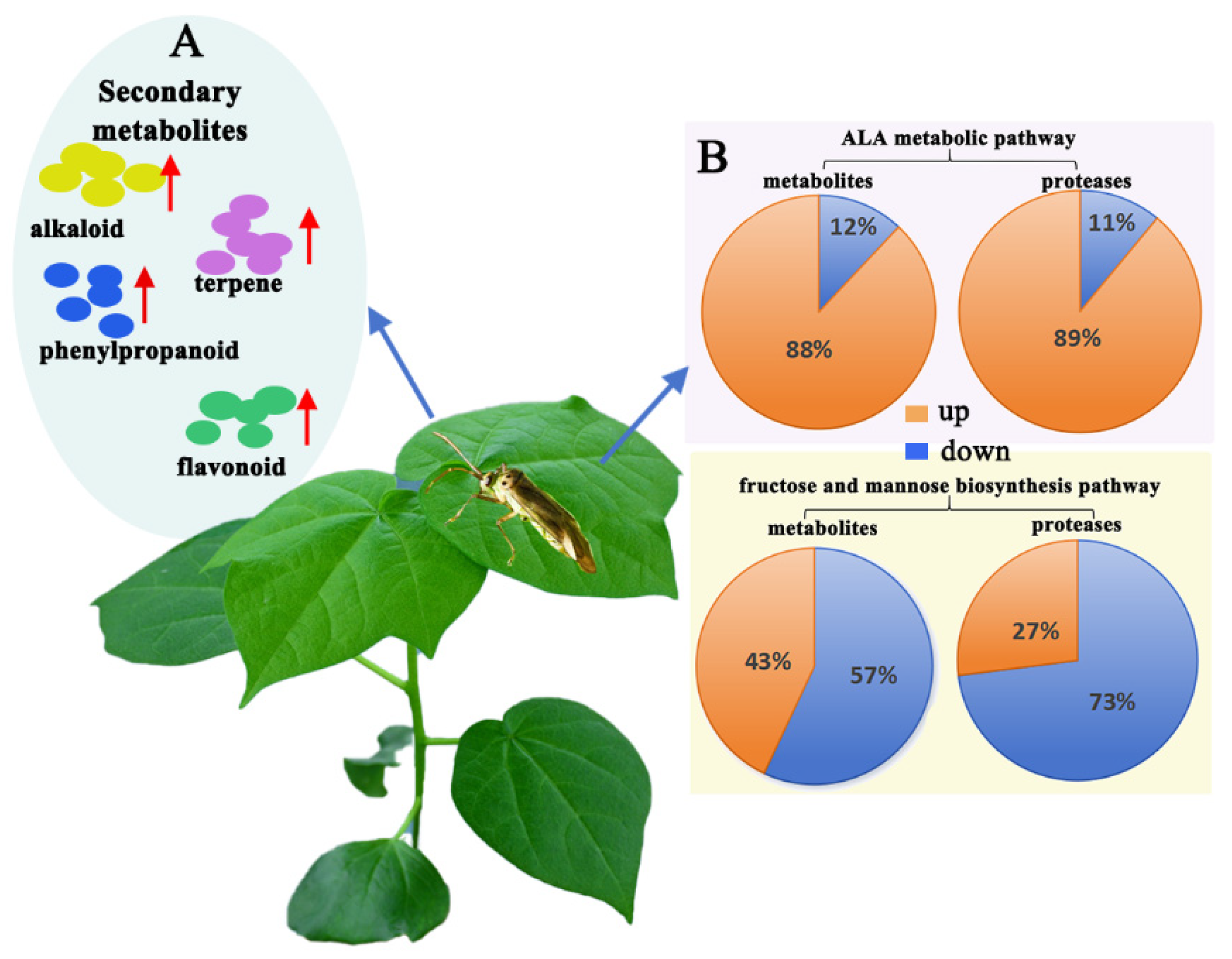
3.3. Integrative Metabolomic–Proteomic Analysis
3.3.1. Integrating Proteomics and Metabolomics for Fructose and Mannose Metabolism Pathway Analysis
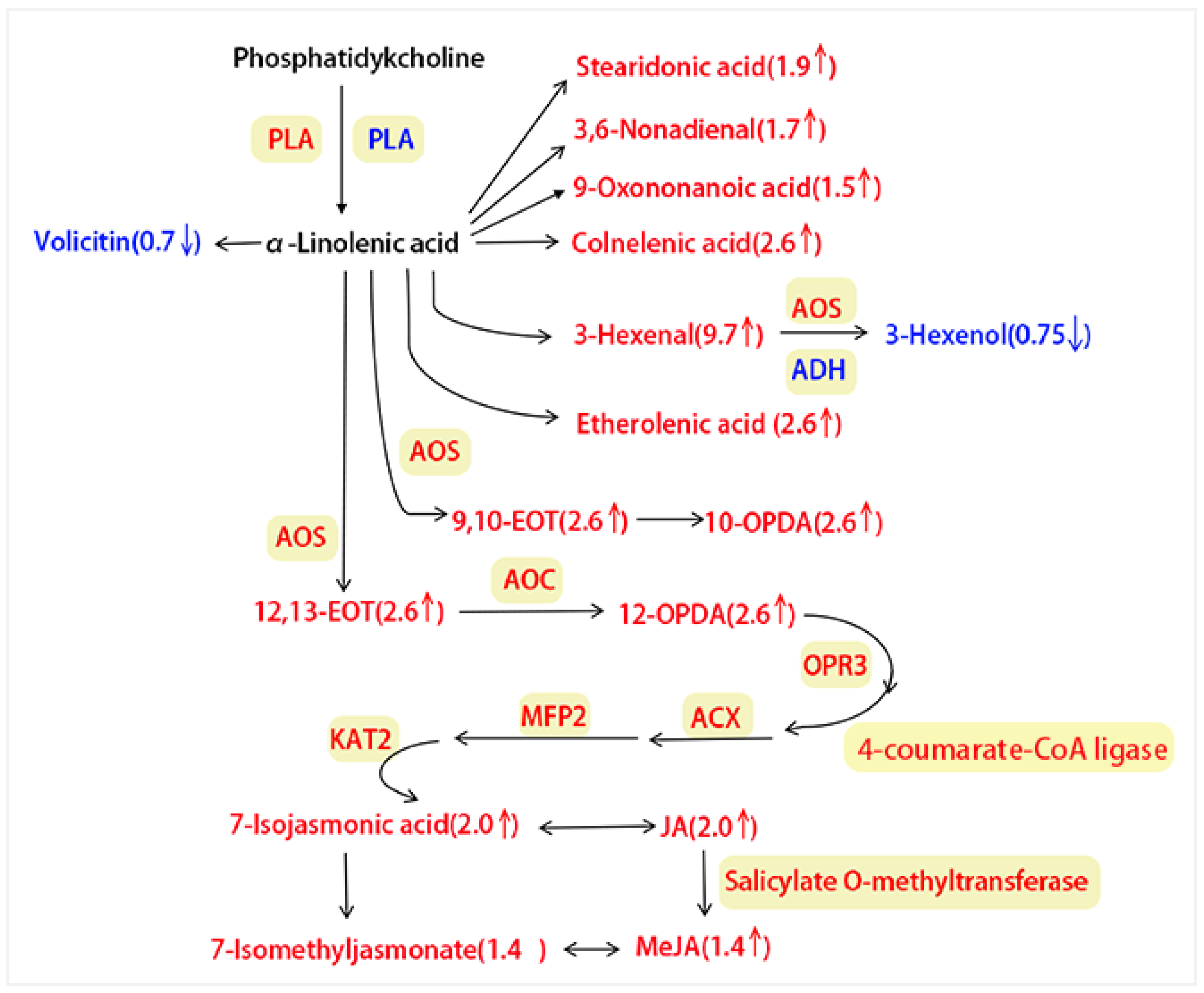
3.3.2. Integrating Proteomics and Metabolomics for ALA Metabolism Pathway Analysis
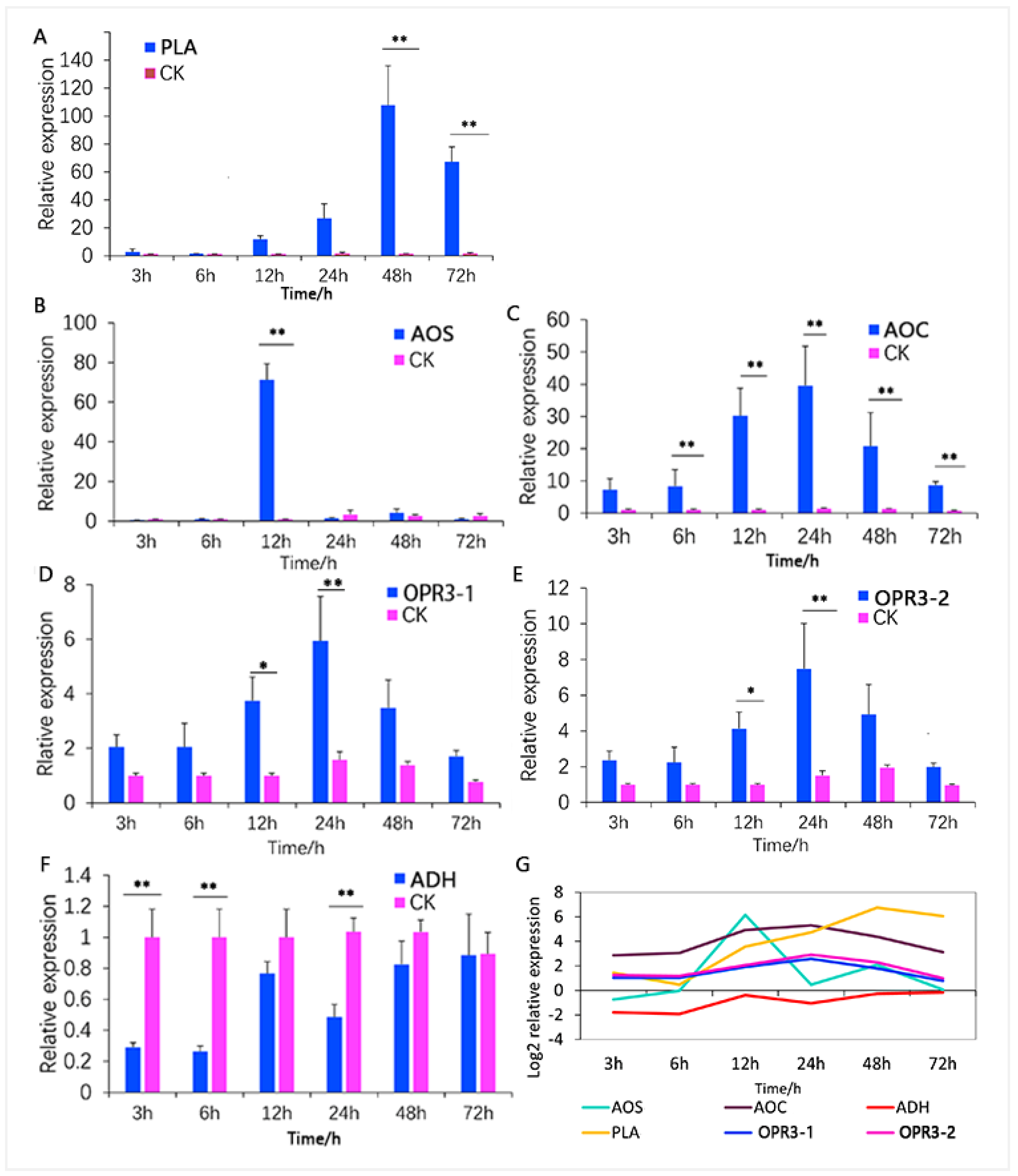
3.4. Analysis of Key ALA Metabolism Pathway Regulators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kessler, A.; Baldwin, I.T. Plant responses to insect herbivory: The emerging molecular analysis. Annu. Rev. Plant Biol. 2002, 53, 299–328. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Baldwin, I.T. New insights into plant responses to the attack from insect herbivores. Annu. Rev. Genet. 2010, 44, 1–24. [Google Scholar] [CrossRef]
- Howe, G.A.; Jander, G. Plant immunity to insect herbivores. Annu. Rev. Plant Biol. 2008, 59, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhou, Q.; Chen, W.; Zhang, G.; He, G.F.; Gu, D.X.; Zhang, W.Q. Involvement of Jasmonate-signaling pathway in the herbivore-induced rice plant defense. Chin. Sci. Bull. 2003, 48, 1982–1987. [Google Scholar] [CrossRef]
- Cao, J.J.; Li, M.Y.; Chen, J.; Liu, P.; Li, Z. Effects of MeJA on Arabidopsis metabolome under endogenous JA deficiency. Sci. Rep. 2016, 6, 37674. [Google Scholar] [CrossRef]
- Townsend, B.J.; Poole, A.; Blake, C.J.; Llewellyn, D.J. Antisense suppression of a (+)-delta-cadinene synthase gene in cotton prevents the induction of this defense response gene during bacterial blight infection but not its constitutive expression. Plant Physiol. 2005, 138, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Song, S. Jasmonates: Biosynthesis, metabolism, and signaling by proteins activating and repressing transcription. J. Exp. Bot. 2017, 68, 1303–1321. [Google Scholar] [CrossRef]
- Chini, A.; Monte, I.; Zamarreño, A.M.; Hamberg, M.; Lassueur, S.; Reymond, P.; Weiss, S.; Stintzi, A.; Schaller, A.; Porzel, A.; et al. An OPR3-independent pathway uses 4,5-didehydrojasmonate for jasmonate synthesis. Nat. Chem. Biol. 2018, 14, 171–178. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.; Cai, X.; Han, Z.; Si, J.; Chen, D. Jasmonate signaling pathway modulates plant defense, growth, and their trade-offs. Int. J. Mol. Sci. 2022, 23, 3945. [Google Scholar] [CrossRef]
- Griffiths, G. Jasmonates: Biosynthesis, perception and signal transduction. Essays Biochem. 2020, 64, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, X.; Llorca, L.C.; Lu, J.; Lou, Y.; Li, R. Role of jasmonate signaling in rice resistance to the leaf folder Cnaphalocrocis medinalis. Plant Mol. Biol. 2022, 109, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.C.; Liu, W.J.; Luo, J.Y.; Wang, L.S.; Zhu, X.Z.; Gao, X.K.; Hua, H.X.; Cui, J.J. Helicoverpa armigera herbivory negatively impacts Aphis gossypii populations via inducible metabolic changes. Pest Manag. Sci. 2022, 78, 2357–2369. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.C.; Qi, J.F.; Zu, H.Y.; Liu, S.T.; Gershenzon, J.; Lou, Y.G.; Baldwin, I.T.; Li, R. Jasmonate-mediated gibberellin catabolism constrains growth during herbivore attack in rice. Plant Cell 2023, 35, 3828–3844. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, T.; Huangfu, J.; Li, R.; Lou, Y. Both allene oxide synthases genes are involved in the biosynthesis of herbivore-induced jasmonic acid and herbivore resistance in rice. Plants 2021, 10, 442. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.X.; Xu, N.; Rui, C.; Fan, Y.P.; Wang, J.; Han, M.; Wang, Q.Q.; Sun, L.Q.; Chen, X.G.; et al. Genome-wide expression analysis of phospholipase A1 (PLA1) gene family suggests phospholipase A1-32 gene responding to abiotic stresses in cotton. Int. J. Biol. Macromol. 2021, 192, 1058–1074. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.T.; Chen, Y.; Feng, A.Y.; Zou, W.H.; Wang, D.J.; Lin, P.X.; Chen, Y.L.; You, C.H.; Que, Y.X.; Su, Y.C. The allene oxide synthase gene family in sugarcane and its involvement in disease resistance. Ind. Crops Prod. 2023, 192, 116–136. [Google Scholar] [CrossRef]
- Available online: https://www.fao.org/newsroom/detail/world-cotton-day-celebrating-the-role-of-cotton-in-global-development-while-calling-for-developing-the-crop-more-sustainably/en (accessed on 12 January 2024).
- Lu, Y.H.; Wu, K.M.; Jiang, Y.Y.; Xia, B.; Li, P.; Feng, H.Q.; Wyckhuys, K.A.; Guo, Y. Mirid bug outbreaks in multiple crops correlated with wide-scale adoption of Bt cotton in China. Science 2010, 328, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.M.; Lu, Y.H.; Feng, H.Q.; Jiang, Y.Y.; Zhao, J.Z. Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin-containing cotton. Science 2008, 321, 1676–1678. [Google Scholar] [CrossRef]
- Lu, Y.H.; Wu, K.M. Mirid bugs in China: Pest status and management strategies. Outlooks Pest Manag. 2011, 22, 248–252. [Google Scholar] [CrossRef]
- Luo, J.; Liu, X.; Liu, L.; Zhang, P.; Chen, L.Z.; Gao, Q.; Ma, W.H.; Chen, L.Z.; Lei, C. De novo analysis of the Adelphocoris suturalis Jakovlev metathoracic scent glands transcriptome and expression patterns of pheromone biosynthesis-related genes. Gene 2014, 551, 271–278. [Google Scholar] [CrossRef]
- Tong, Y.J.; Wu, K.M.; Lu, Y.H.; Gao, X.W. Pathogenicity of Beauveria spp. strains to three species of mirids, Apolygus lucorum, Adelphocoris suturalis and Adelphocoris lineolatus. Agric. Food Sci. 2010, 37, 172176. Available online: https://api.semanticscholar.org/CorpusID:90497852 (accessed on 2 March 2018).
- Luo, J.; Liang, S.j.; Li, J.Y.; Xu, Z.P.; Li, L.; Zhu, B.Q.; Li, Z.; Lei, C.L.; Lindsey, K.; Chen, L.Z.; et al. A transgenic strategy for controlling plant bugs (Adelphocoris suturalis) through expression of doublestranded RNA homologous to fatty acyl-coenzyme a reductase in cotton. New Phytol. 2017, 215, 1173–1185. [Google Scholar] [CrossRef]
- Ma, M.Q.; Guo, L.B.; Tu, C.J.; Wang, A.L.; Xu, L.T.; Luo, J. Comparative analysis of Adelphocoris suturalis Jakovlev (Hemiptera: Miridae) immune responses to fungal and bacterial pathogens. Front. Physiol. 2021, 18, 646721. [Google Scholar] [CrossRef]
- Xue, H.; Zhu, X.Z.; Wang, L.; Zhang, K.X.; Li, D.Y.; Ji, J.C.; Niu, L.; Wu, C.C.; Gao, X.K.; Luo, J.Y.; et al. Gut bacterial diversity in different life cycle stages of Adelphocoris suturalis (Hemiptera: Miridae). Front. Microbiol. 2021, 12, 670383. [Google Scholar] [CrossRef]
- Lu, Y.H.; Jiao, Z.; Li, G.; Wyckhuys, K.A.G.; Wu, K.M. Comparative overwintering host range of three Adelphocoris species (Hemiptera: Miridae) in northern China. Crop Protect. 2011, 30, 1455–1460. [Google Scholar] [CrossRef]
- Luo, J.; Li, Z.; Ma, C.; Zhang, Z.; Hull, J.J.; Lei, C.; Jin, S.; Chen, L. Knockdown of a metathoracic scent gland desaturase enhances the production of (E)-4-oxo-2-hexenal and suppresses female sexual attractiveness in the plant bug Adelphocoris suturalis. Insect Mol. Biol. 2017, 26, 642–653. [Google Scholar] [CrossRef]
- Xue, H.; Huang, X.X.; Chang, G.F.; Ma, W.H.; Hull, J.J.; Chen, L.Z. Reproductive capacity in Adelphocoris suturalis (Hemiptera: Miridae) is regulated by the insulin signaling pathway. Pestic. Biochem. Physiol. 2022, 187, 105195. [Google Scholar] [CrossRef]
- Qin, S.; Zhu, B.; Huang, X.; Hull, J.J.; Chen, L.; Luo, J. Functional Role of AsAP in the Reproduction of Adelphocoris suturalis (Hemiptera: Miridae). Insects 2022, 13, 755. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; Auria, J.D.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.W.; Wang, G.; Baek, S.J.; Shen, R.F. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF. J. Proteome Res. 2006, 5, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Hou, Y.; Zhou, X.Y.; Chen, Q.M.; Guo, C.; Xia, Q.Y.; Zhang, Y.; Zhao, P. Integrative proteomics and metabolomics analysis of insect larva brain: Novel insights into the molecular mechanism of insect wandering behavior. J. Proteome Res. 2016, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Liu, C.W.; Huang, X.H.; Zhou, X.; Zhuo, J.C.; Zhang, C.X.; Bao, Y.Y. Screening and functional analyses of Nilaparvata lugens salivary proteome. J. Proteome Res. 2016, 15, 1883–1896. [Google Scholar] [CrossRef]
- Keeling, C.I.; Li, M.; Sandhu, H.K.; Henderson, H.; Yuen, M.M.; Bohlmann, J. Quantitative metabolome, proteome and transcriptome analysis of midgut and fat body tissues in the mountain pine beetle, Dendroctonus ponderosae Hopkins, and insights into pheromone biosynthesis. Insect Biochem. Mol. Biol. 2016, 70, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Wang, J.; Hao, K.; Whitman, D.W.; Fan, Y.; Cao, G.; Zhang, Z. Transcriptomic and proteomic analysis of pre-diapause and non-diapause eggs of migratory locust, Locusta migratoria L. (Orthoptera: Acridoidea). Sci. Rep. 2015, 5, 11402. [Google Scholar] [CrossRef]
- Liu, J.; Pang, C.Y.; Wei, H.L.; Song, M.Z.; Meng, Y.Y.; Ma, J.H.; Fan, S.L.; Yu, S.X. Data for proteomic profiling of Anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). Data Brief 2015, 4, 500–509. [Google Scholar] [CrossRef]
- Liu, Q.S.; Wang, X.Y.; Tzin, V.; Romeis, J.; Peng, Y.F.; Li, Y.H. Combined transcriptome and metabolome analyses to understand the dynamic responses of rice plants to attack by the rice stem borer Chilo suppressalis (Lepidoptera: Crambidae). BMC Plant Biol. 2016, 16, 259. [Google Scholar] [CrossRef]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-based metabolomics. Mol. Biosyst. 2012, 2, 470–481. [Google Scholar] [CrossRef]
- Maiväli, Ü. Chapter 4—Data and Evidence. In Interpreting Biomedical Science; Academic Press: Cambridge, MA, USA, 2015; pp. 159–221. ISBN 9780124186897. [Google Scholar] [CrossRef]
- Zhou, C.X.; Zhou, D.H.; Elsheikha, H.M.; Zhao, Y.; Suo, X.; Zhu, X.Q. Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Sci. Rep. 2016, 6, 19557. [Google Scholar] [CrossRef]
- Zhao, S.Y.; Liu, Z.L.; Shu, Y.S.; Wang, M.L.; He, D.; Song, Z.Q.; Zeng, H.L.; Ning, Z.C.; Lu, C.; Lu, A.P.; et al. Chemotaxonomic classification applied to the identification of two closely-related citrus TCMs using UPLC-Q-TOF-MS-based metabolomics. Molecules 2017, 22, 1721. [Google Scholar] [CrossRef]
- Chen, T.T.; Zhang, L.; Shang, H.; Liu, S.; Peng, J.; Gong, W.; Shi, Y.; Zhang, S.; Li, J.; Gong, J.; et al. iTRAQ-Based quantitative proteomic analysis of cotton roots and leaves reveals pathways associated with salt stress. PLoS ONE 2016, 11, e0148487. [Google Scholar] [CrossRef]
- Saravanan, R.S.; Rose, J.K. A critical evaluation of sample extraction techniques for enhanced proteomic analysis of recalcitrant plant tissues. Proteomics 2004, 4, 2522–2532. [Google Scholar] [CrossRef]
- Petricka, J.J.; Schauer, M.A.; Megraw, M.; Breakfield, N.W.; Thompson, J.W.; Georgiev, S.; Soderblom, E.J.; Ohler, U.; Moseley, M.A.; Grossniklaus, U.; et al. The protein expression landscape of the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2012, 109, 6811–6818. [Google Scholar] [CrossRef]
- Kruger, N.J. The Bradford method for protein quantitation. Methods Mol. Biol. 1994, 32, 9–15. [Google Scholar] [CrossRef]
- Liu, J.F.; Xing, J.Y.; Fang, J.; Ai, P.F.; Cheng, Y.Q. New insight into ovary abortion during ovary development of hazelnut through a combined proteomic and transcriptomic analysis. Sci. Hortic. 2018, 234, 36–48. [Google Scholar] [CrossRef]
- Chu, P.; Yan, G.X.; Yang, Q.; Zhai, L.N.; Zhang, C.; Zhang, F.Q.; Guan, R.Z. iTRAQ-based quantitative proteomics analysis of Brassica napus leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 2015, 113, 244–259. [Google Scholar] [CrossRef]
- Zhu, M.M.; Dai, S.J.; McClung, S.; Yan, X.F.; Chen, S.X. Functional differentiation of Brassica napus guard cells and mesophyll cells revealed by comparative proteomics. Mol. Cell. Proteomics 2009, 8, 752–766. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Pang, Q.Y.; He, Y.; Zhu, N.; Branstrom, I.; Yan, X.F.; Chen, S. Proteomics and metabolomics of Arabidopsis responses to perturbation of glucosinolate biosynthesis. Mol. Plant 2012, 5, 1138–1150. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time Quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.L.; Zhang, B.H. Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.). Gene 2013, 530, 44–50. [Google Scholar] [CrossRef]
- Jonsson, P.; Bruce, S.J.; Moritz, T.; Trygg, J.; Sjöström, M.; Plumb, R.; Granger, J.; Maibaum, E.; Nicholson, J.K.; Holmes, E.; et al. Extraction, interpretation and validation of information for comparing samples in metabolic LC/MS data sets. Analyst 2005, 130, 701–707. [Google Scholar] [CrossRef]
- Chiozza, M.V.; O’Neal, M.E.; MacIntosh, G.C. Constitutive and induced differential accumulation of amino acid in leaves of susceptible and resistant soybean plants in response to the soybean aphid (Hemiptera: Aphididae). Environ. Entomol. 2010, 39, 856–864. [Google Scholar] [CrossRef]
- Gols, R. Direct and indirect chemical defences against insects in a multitrophic framework. Plant Cell Environ. 2014, 7, 1741–1752. [Google Scholar] [CrossRef]
- Erb, M.; Reymond, P. Molecular interactions between plants and insect herbivores. Annu. Rev. Plant Biol. 2019, 29, 527–557. [Google Scholar] [CrossRef]
- Fürstenberg-Hägg, J.; Zagrobelny, M.; Bak, S. Plant defense against insect herbivores. Int. J. Mol. Sci. 2013, 14, 10242–10297. [Google Scholar] [CrossRef]
- Beran, F.; Petschenka, G. Sequestration of plant defense compounds by insects: From mechanisms to insect-plant coevolution. Annu. Rev. Entomol. 2022, 67, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.; Heuskin, S.; Delaplace, P.; Francis, F.; Lognay, G. VOC emissions and protein expression mediated by the interactions between herbivorous insects and Arabidopsis plant. Biotechnol. Agron. Soc. Environ. 2014, 18, 455–464. [Google Scholar]
- Bede, J.C.; Musser, R.O.; Felton, G.W.; Korth, K.L. Caterpillar herbivory and salivary enzymes decrease transcript levels of Medicago truncatula genes encoding early enzymes in terpenoid biosynthesis. Plant Mol. Biol. 2006, 60, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, G. Plants recognize. herbivorous insects by complex signalling networks. Annu. Plant Rev. 2014, 47, 1–35. [Google Scholar] [CrossRef]
- Divekar, P.A.; Narayana, S.; Divekar, B.A.; Kumar, R.; Gadratagi, B.G.; Ray, A.; Singh, A.K.; Rani, V.; Singh, V.; Singh, A.K.; et al. Plant secondary metabolites as defense tools against herbivores for sustainable crop protection. Int. J. Mol. Sci. 2022, 23, 2690. [Google Scholar] [CrossRef] [PubMed]
- Hogenhout, S.A.; Bos, J.I. Effector proteins that modulate plant--insect interactions. Curr. Opin. Plant. Biol. 2011, 14, 422–428. [Google Scholar] [CrossRef]
- Stam, J.M.; Kroes, A.; Li, Y.; Gols, R.; van Loon, J.J.; Poelman, E.H.; Dicke, M. Plant interactions with multiple insect herbivores: From community to genes. Annu. Rev. Plant. Biol. 2014, 65, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Alborn, H.T.; Turlings, T.C.J.; Jones, T.H.; Stenhagen, G.; Loughrin, J.H.; Tumlinson, J.H. An Elicitor of plant volatiles from beet armyworm oral secretion. Science 1997, 276, 945–949. [Google Scholar] [CrossRef]
- Chen, C.Y.; Liu, Y.Q.; Song, W.M.; Chen, D.Y.; Chen, F.Y.; Chen, X.Y.; Chen, Z.W.; Ge, S.X.; Wang, C.Z.; Zhan, S.; et al. An effector from cotton bollworm oral secretion impairs host plant defense signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 14331–14338. [Google Scholar] [CrossRef]
- Wang, Y.; Mostafa, S.; Zeng, W.; Jin, B. Function and mechanism of jasmonic acid in plant responses to abiotic and biotic stresses. Int. J. Mol. Sci. 2021, 22, 8568. [Google Scholar] [CrossRef]
- Erb, M.; Kliebenstein, D.J. Plant secondary metabolites as defenses, regulators, and primary metabolites: The blurred functional trichotomy. Plant Physiol. 2020, 184, 39–52. [Google Scholar] [CrossRef]
- Haukioja, E.; Neuvonen, S. Induced long-term resistance of birth foliage against defoliators: Defensive or incidental? Ecology 1985, 66, 1303–1308. [Google Scholar] [CrossRef]
- Hunter, M.D.; Schultz, J.C. Fetilization mitigates chemical induction and herbivore responses within damaged oak trees. Ecology 1995, 76, 1226–1232. [Google Scholar] [CrossRef]
- Ninkuu, V.; Zhang, L.; Yan, J.P.; Fu, Z.C.; Yang, T.F.; Zeng, H.M. Biochemistry of terpenes andrecent advances in plant protection. Int. J. Mol. Sci. 2021, 22, 5710. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pathway_ID | KEGG Pathway Name | Total | Down | Up |
|---|---|---|---|---|---|
| 1 | map00909 | Sesquiterpenoid and triterpenoid biosynthesis | 40 | 0 | 40 |
| 2 | map00590 | Arachidonic acid metabolism | 28 | 7 | 21 |
| 3 | map00380 | Tryptophan metabolism | 23 | 5 | 18 |
| 4 | map00904 | Diterpenoid biosynthesis | 20 | 6 | 14 |
| 5 | map00950 | Isoquinoline alkaloid biosynthesis | 19 | 5 | 14 |
| 6 | map00960 | Tropane, piperidine and pyridine alkaloid biosynthesis | 19 | 2 | 17 |
| 7 | map00940 | Phenylpropanoid biosynthesis | 18 | 2 | 16 |
| 8 | map00350 | Tyrosine metabolism | 17 | 2 | 15 |
| 9 | map00592 | alpha-Linolenic acid metabolism | 16 | 2 | 14 |
| 10 | map00902 | Monoterpenoid biosynthesis | 15 | 5 | 10 |
| 11 | map00051 | Fructose and mannose metabolism | 14 | 8 | 6 |
| 12 | map00360 | Phenylalanine metabolism | 14 | 2 | 12 |
| 13 | map00903 | Limonene and pinene degradation | 14 | 3 | 9 |
| 14 | map00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 13 | 3 | 10 |
| 15 | map00330 | Arginine and proline metabolism | 11 | 2 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Zheng, S.; Ma, C.; Gao, X.; Ji, J.; Luo, J.; Hua, H.; Cui, J. Integrated Omics Analysis Reveals Key Pathways in Cotton Defense against Mirid Bug (Adelphocoris suturalis Jakovlev) Feeding. Insects 2024, 15, 254. https://doi.org/10.3390/insects15040254
Lu H, Zheng S, Ma C, Gao X, Ji J, Luo J, Hua H, Cui J. Integrated Omics Analysis Reveals Key Pathways in Cotton Defense against Mirid Bug (Adelphocoris suturalis Jakovlev) Feeding. Insects. 2024; 15(4):254. https://doi.org/10.3390/insects15040254
Chicago/Turabian StyleLu, Hui, Shuaichao Zheng, Chao Ma, Xueke Gao, Jichao Ji, Junyu Luo, Hongxia Hua, and Jinjie Cui. 2024. "Integrated Omics Analysis Reveals Key Pathways in Cotton Defense against Mirid Bug (Adelphocoris suturalis Jakovlev) Feeding" Insects 15, no. 4: 254. https://doi.org/10.3390/insects15040254
APA StyleLu, H., Zheng, S., Ma, C., Gao, X., Ji, J., Luo, J., Hua, H., & Cui, J. (2024). Integrated Omics Analysis Reveals Key Pathways in Cotton Defense against Mirid Bug (Adelphocoris suturalis Jakovlev) Feeding. Insects, 15(4), 254. https://doi.org/10.3390/insects15040254